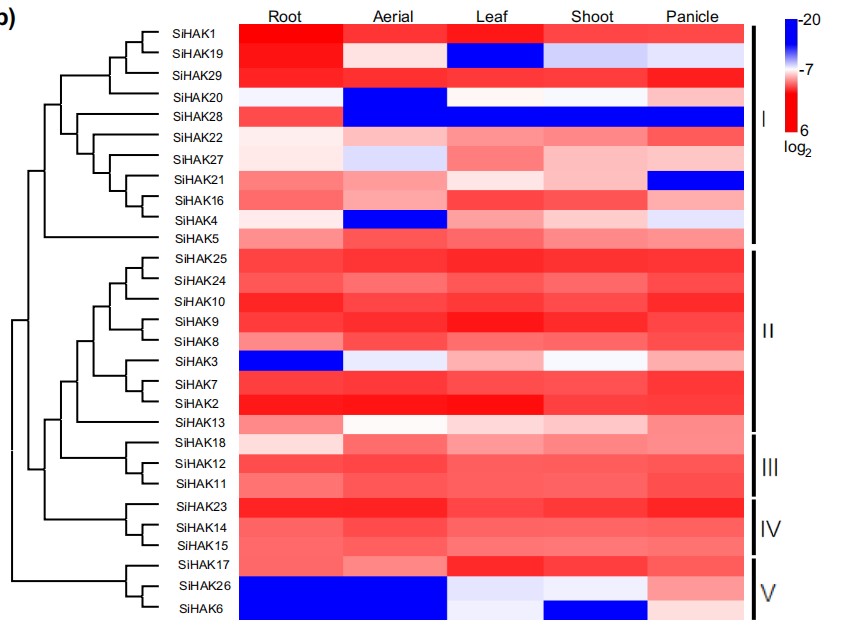
请问老师是不是过程 和全基因组关联分析一样,和物种的全基因组数据bwa,samtools生成bam文件,再cuffdiff或者htseq-count进行分析,用edger分析得到gene的各种log2FoldChange pval PADJ等数据,再将筛选的处理的家族geneid挑出来进行热图绘制吗?
请问老师是不是过程 和全基因组关联分析一样,和物种的全基因组数据bwa,samtools生成bam文件,再cuffdiff或者htseq-count进行分析,用edger分析得到gene的各种log2FoldChange pval PADJ等数据,再将筛选的处理的家族geneid挑出来进行热图绘制吗?
如果只有原始数据fastq文件或者NCBI上下载的sra数据,就需要自己手动比对,定量分析。如果没有实验室没有服务器,自己得笔记本分析原始测序数据很吃力,不建议分析。
有兴趣可以看这门课程:二代测序转录组数据自主分析
我们公司也也可以分析转录组数据,可以联系我们,18510686772
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

同学你好,能不能给一下视频购买链接啊?非常想要!!感谢感谢