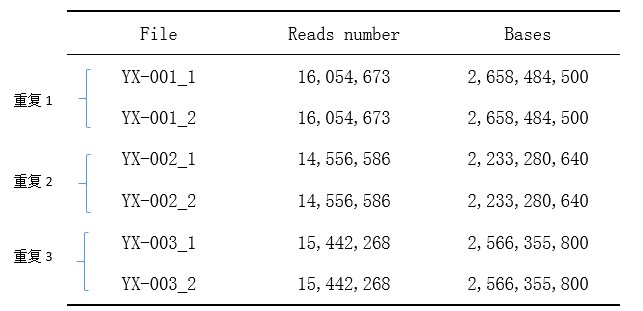
看你的数据,应该是一个样品取了三个生物学重复(重复1,重复2,重复3),每一个重复里面的(1和2)应该是双端测序得到的read1和read2两个文件的统计结果,因为后面readnumber和bases数量(两个文件如 YX-001_1 和YX-001_2 )是一样的。
1. YX-001 的bases数可以 YX-001_1 和YX-001_2 相加; read数量你直接用16,054,673就行,说明一下是一对记为1条reads,也可以相加,说按单端计数;
2,所有的read数量也就是测序数据量,你可以相加,文章结果总结可以说一下,一共测了多少read,多少数据量base;
3. 双端测序read1和read2的read数量一定是相等的,不相等说明文件有错误;
更多测序数据说明见:《illumina二代测序原理及fastq视频课程》;
