发现
问答
发起
提问
文章
文章
更多
专家
讲堂
话题
财富榜
商城
Toggle navigation
首页
(current)
问答
文章
视频课程
话题
商城
搜索
登录
注册
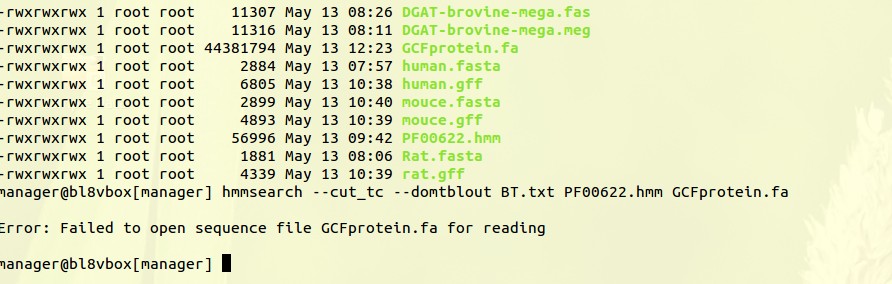
你好,我在利用虚拟机做hmmersearch时遇见这种问题,该如何解决
hmmsearch
0 条评论
分类:
基因家族分析
请先
登录
后评论
默认排序
时间排序
1 个回答
omicsgene
- 生物信息
2020-05-14 10:41
擅长:重测序,遗传进化,转录组,GWAS
输入的那个fasta文件有问题吧,你检查一下,或者文件路径写错了。
Linux没基础应该学习:
linux系统使用
请先
登录
后评论
×
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
您需要登录后才可以回答问题,
登录
或者
注册
关注
1
关注
收藏
0
收藏,
3399
浏览
伶俐
提出于 2020-05-13 22:45
相似问题
老师,请问下两次hmmsearch的结果一样正常吗?
1 回答
为什么我的bioLinux打不开.fa的文件呢
1 回答
阈值太大怎么办
1 回答
老师,建立hmmbuild模型后,再用hmmsearch搜索,搜索结果比第一次的还多的情况是正常的吗?
1 回答
请问HMM搜索,对于无蛋白文件的基因组,可以对实验室自有的转录组序列或数据做筛选吗?
1 回答
×
发送私信
发给:
内容:
×
举报此文章
垃圾广告信息:
广告、推广、测试等内容
违规内容:
色情、暴力、血腥、敏感信息等内容
不友善内容:
人身攻击、挑衅辱骂、恶意行为
其他原因:
请补充说明
举报原因: