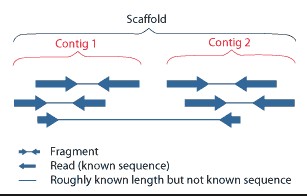
基因组de novo测序,通过reads拼接获得Contigs后,再通过大片段文库(如3Kb、6Kb、10Kb、20Kb)两端的序列。来确定一些Contig之间的顺序关系,这些先后顺序已知的Contigs组成Scaffold。
2 个回答
擅长:重测序,遗传进化,转录组,GWAS
A contig is a contiguous length of genomic sequence.
A scaffold is composed of contigs and gaps. Gap length can be guessed by incorporating information from paired ends or mate pairs
What is a Scaffold? |
A scaffold is a portion of the genome sequence reconstructed from end-sequenced whole-genome shotgun clones. Scaffolds are composed of contigs and gaps. A contig is a contiguous length of genomic sequence in which the order of bases is known to a high confidence level. Gaps occur where reads from the two sequenced ends of at least one fragment overlap with other reads in two different contigs (as long as the arrangement is otherwise consistent with the contigs being adjacent). Since the lengths of the fragments are roughly known, the number of bases between contigs can be estimated.
The goal of whole-genome shotgun assembly is to represent each genomic sequence in one scaffold; however, this is not always possible. One chromosome may be represented by many scaffolds (e.g., Chlamydomonas reinhardtii) or just a single scaffold (e.g., Human chromosome 19), depending on how completely the genome can be reconstructed, or assembled, from the available reads. The relative locations of scaffolds in the genome are unknown. Scaffolds are normally numbered approximately from largest to smallest. Some scaffolds may ultimately be filtered out of the assembly, resulting in skipped scaffold numbers. In some cases, scaffolds can overlap. For example, in polymorphic genomes, regions with a high density of allelic differences between haplotypes may be split into separate sets of scaffolds, each representing one allele. Thus, a sequence that exists in only one location in the genome may appear on more than one scaffold. Gaps are shown in the Genome Viewer as red lines or rectangles in the scaffold track (viewed in "full" mode). Contigs are shown in black. In FASTA sequences, gaps are represented by a series of Ns.
|
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读、
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
