看脚本应该是没问题的,可能是你的输入ID列表文件有问题,windows中的换行符和linux的不兼容,建议编辑的时候设置一下UTF-8
https://www.omicsclass.com/article/395 再重新输入试试;
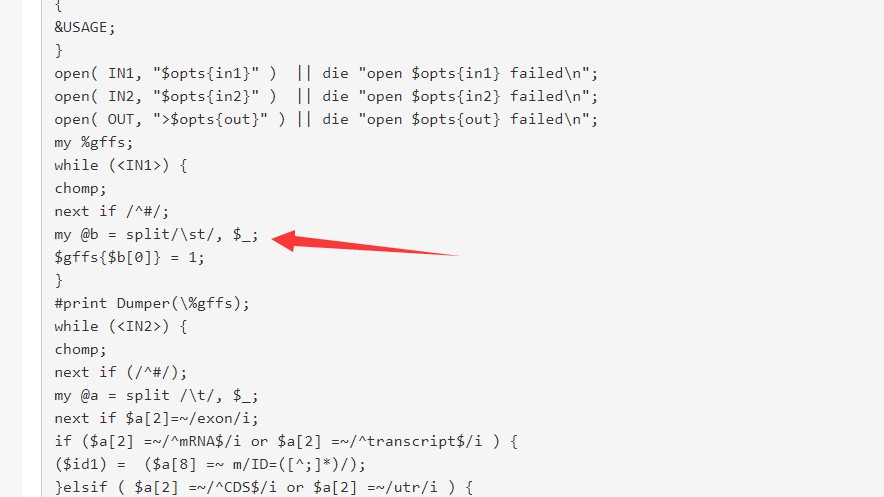
这个地方改成:
my @b = split/\s+/, $_;
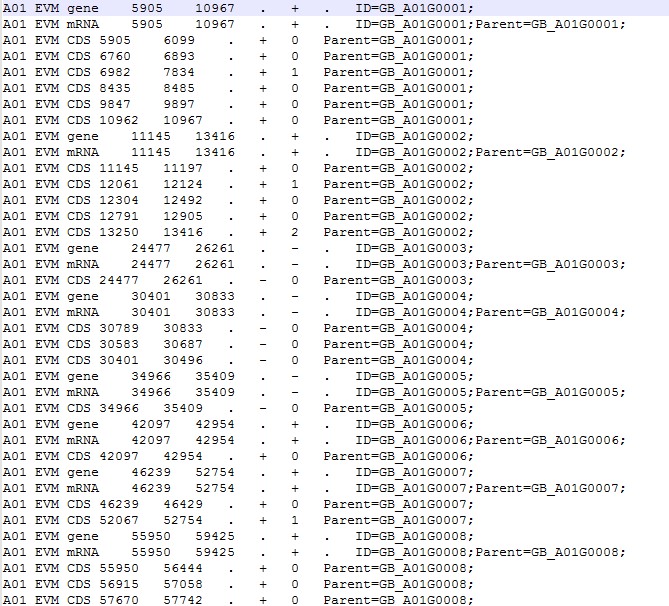
这是我的GFF文件
下图是运行程序,运行过程中没报错,但结果文件是空的

这是ID
这是脚本文件
use Getopt::Long;
my %opts;
use Data::Dumper;
GetOptions( \%opts, "in1=s", "in2=s", "out=s", "h" );
if ( !defined( $opts{in1} )
|| !defined( $opts{in2} )
|| !defined( $opts{out} )
|| defined( $opts{h} ) )
{
&USAGE;
}
open( IN1, "$opts{in1}" ) || die "open $opts{in1} failed\n";
open( IN2, "$opts{in2}" ) || die "open $opts{in2} failed\n";
open( OUT, ">$opts{out}" ) || die "open $opts{out} failed\n";
my %gffs;
while (<IN1>) {
chomp;
next if /^#/;
my @b = split/\st/, $_;
$gffs{$b[0]} = 1;
}
#print Dumper(\%gffs);
while (<IN2>) {
chomp;
next if (/^#/);
my @a = split /\t/, $_;
next if $a[2]=~/exon/i;
if ($a[2] =~/^mRNA$/i or $a[2] =~/^transcript$/i ) {
($id1) = ($a[8] =~ m/ID=([^;]*)/);
}elsif ( $a[2] =~/^CDS$/i or $a[2] =~/utr/i ) {
($id1) = ($a[8] =~ m/Parent=([^;]*)/);
}else{
next;
}
if ( exists $gffs{$id1} ) {
print OUT "$_\n";
}
}
close OUT;
close IN1;
close IN2;
sub USAGE {
print "usage: perl $0 -in1 mRNA_id.txt -in2 genome.gff3 -out gene_location.txt ";
exit;
}
看脚本应该是没问题的,可能是你的输入ID列表文件有问题,windows中的换行符和linux的不兼容,建议编辑的时候设置一下UTF-8
https://www.omicsclass.com/article/395 再重新输入试试;
这个地方改成:
my @b = split/\s+/, $_;
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
