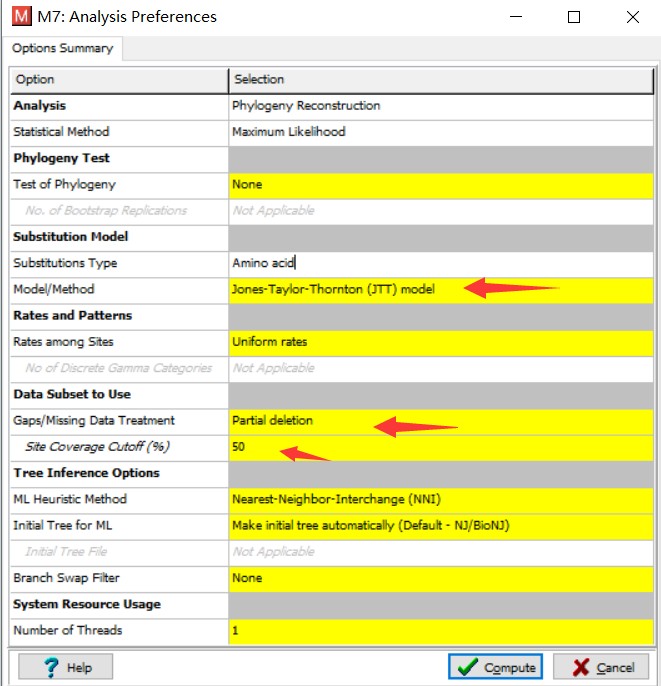
理论上应该尝试各种模型,根据检验结果选择最合适的模型进行计算。但在实际操作中,可先尝试选用较简单的距离模型,比如 p-distance。第三个参数是 Gap/Missing Data Treatment。大多数建树方法会要求删除多序列比对中含有空位的列。但是根据遗传距离度量方法的不同,删除原则也不同。如果是以序列间不同残基的个数来度量遗传距离的话,这里需要选择 Complete deletion(全部删除)。如果是其他方法,比如这里选用的 NJ 方法,可以选择 Partial deletion(部分删除)。删除程度定在 50%,即,保留一半含有空位的列。
选择进化距离模型是构建进化树的基础。DNA 分子中基因的进化距离是通过对核苷酸替代数进行估计获得的,要估计核苷酸替代数,就必须应用核苷酸替代的数学模型。举个例子:在DNA中,碱基之间存在四种转换(A→G,G→A,C→T,T→C)和颠换(A→C,A→T,C→G,G→T),通常情况下转换频率比颠换频率高。
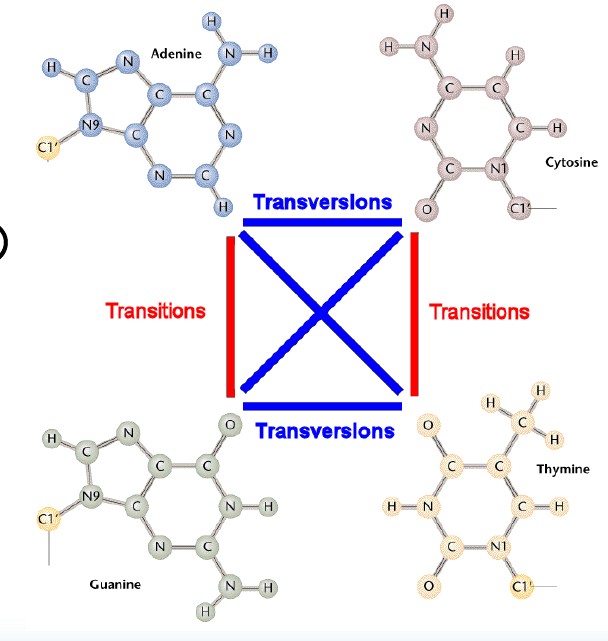
这些偏向会影响两个序列之间的预计分歧。由于DNA 序列的进化、演化比较复杂,因此许多学者提出了不同的核苷酸替代模型。当今常用的核苷酸替代模型有JC69 、K80 、F81、TN93,HKY和GTR 模型等。当然氨基酸也有很多替换模型。这些不同的替换模型确定了不同的进化距离和不同的系统发育树。但实际的生物进化历史是唯一的,我们并不能从这么多的模型中确定真实的核苷酸替代过程是依照哪种模型发生的。理论上应该尝试各种模型,根据检验结果选择最合适的模型进行计算。
在系统发育分析中,最大似然法(ML)和贝叶斯法(BI)是对替换模型非常敏感的两种算法,因此利用ML法或BI法重建系统发育树前,核苷酸替换模型的选择是必不可少的过程;最大简约法(Maximum parsimony)不依赖任何进化模型,因此通过最大简约法构建系统发育树时,不需要此步骤。一般来讲,如果模型合适,ML的效果较好。对近缘序列,有人喜欢MP,因为用的假设最少。MP一般不用在远缘序列上,这时一般用NJ或ML。对相似度很低的序列,NJ往往出现Long-branch attraction(LBA,长枝吸引现象),有时严重干扰进化树的构建。贝叶斯的方法则太慢。对于各种方法构建分子进化树的准确性,一篇综述(Hall BG. Mol Biol Evol 2005, 22(3):792-802)认为贝叶斯的方法最好,其次是ML,然后是MP。其实如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大。
