请将问题说明白,我这才好给你分析原因,因为每个人输入的文件都有差异,你不给我看,我这猜测怎么解决问题呢?
参考一下别人怎么 提交问题:https://www.omicsclass.com/question/242
请将问题说明白,我这才好给你分析原因,因为每个人输入的文件都有差异,你不给我看,我这猜测怎么解决问题呢?
参考一下别人怎么 提交问题:https://www.omicsclass.com/question/242
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

老师,是这样的,我的物种是川桑(Morus notabilis),没有收录在在Ensembl数据库,所以我是在川桑数据库下载的genome,gene,cds及protein。如下图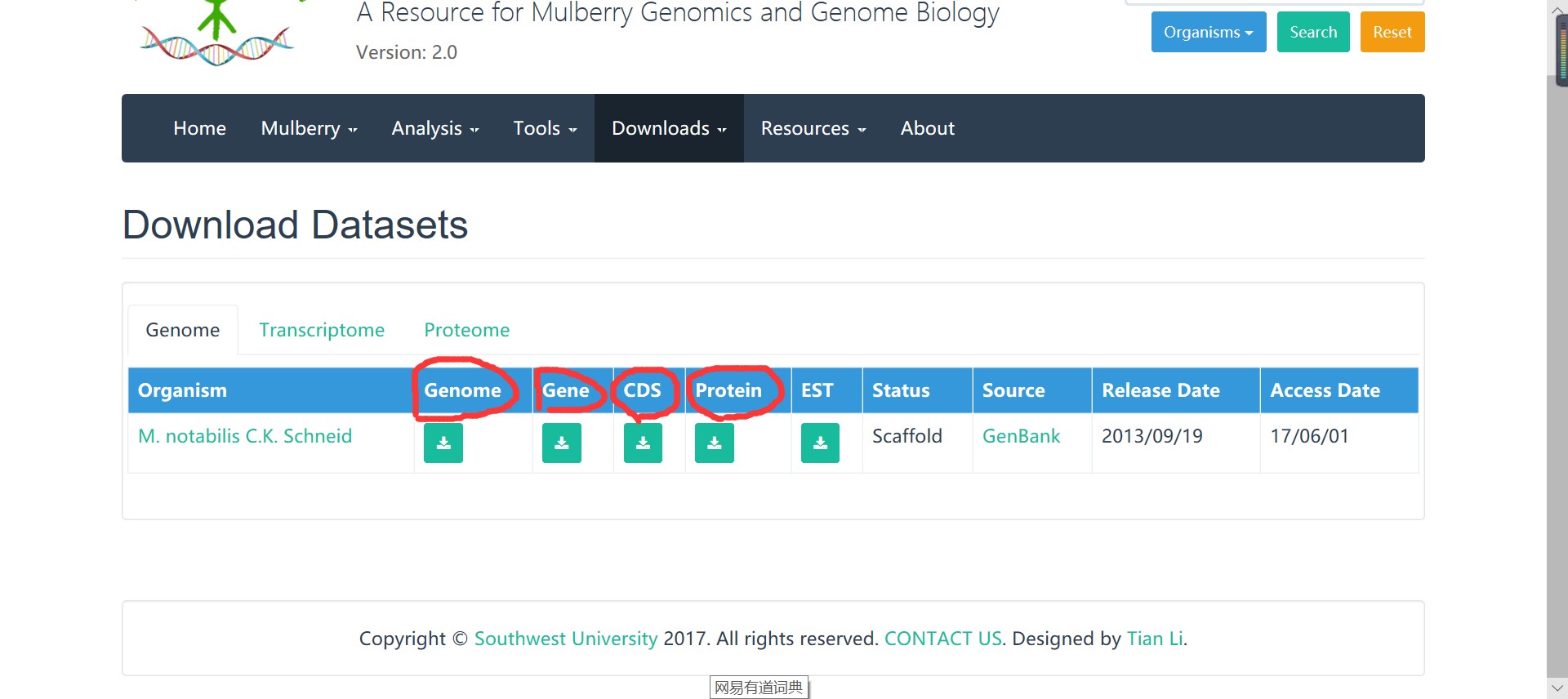
然后在NCBI上下载了川桑的GFF注释文件,如下图
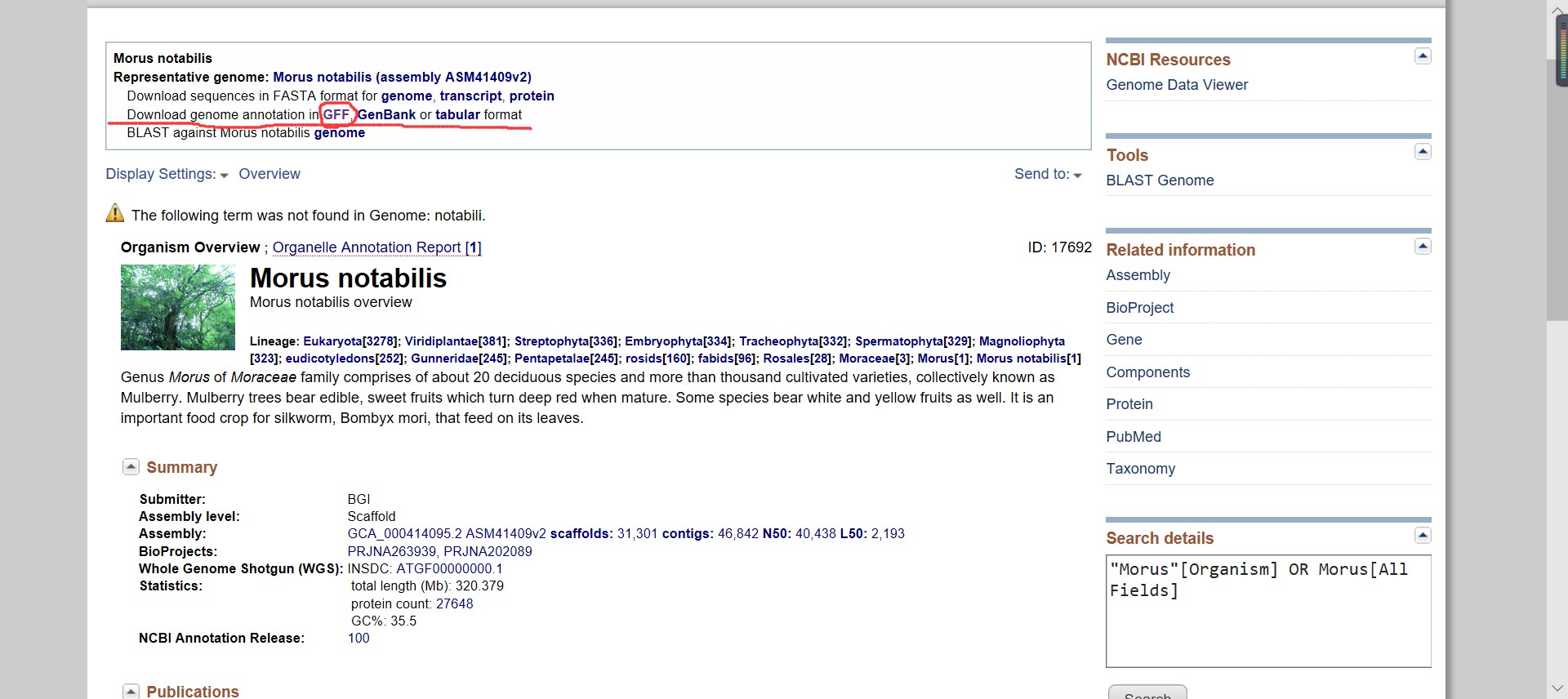
老师,下图是我根据视频学习找到的基因ID 然后运行perl脚本get_gene_weizhi,得到gene_weizhi.txt文件,如下图
然后运行perl脚本get_gene_weizhi,得到gene_weizhi.txt文件,如下图
老师,这儿我补充一点,川桑基因组数据库没有基因在哪条染色体上的信息,所以这里的结果就没有第二列出现基因ID的染色体位置,图中应该是基因在某个片段上的信息,我猜想不管是在染色体上还是在染色体片段上,应该都能找到上游1500位置的序列,所以我就接着往下走,运行Perl脚本tiquxulie,这里perl脚本后面跟的是genome,然后界面上就会出现如下信息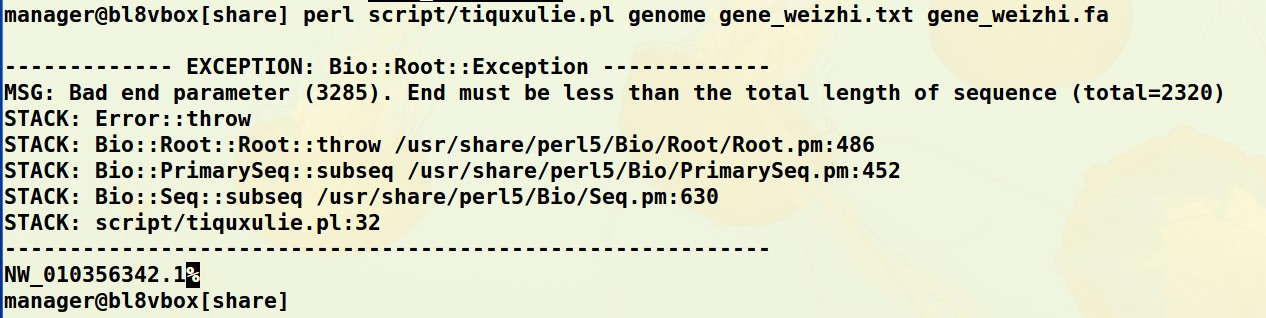
然后查看gene_weizhi.fa,得到如下结构,只有一列短的波浪线
然后我又运行这个脚本,只是perl脚本后面跟的是morus_notabilis.gene.fa,然后出现提示信息
然后查看Ggene_weizhi.fa,跟上面一样的结果
老师,我这个是怎么回事儿呀,我又是初入这个领域,才学疏浅,望老师指导!