提取不出序列,你看看你基因的位置信息里面的第一列里面的序列ID,和你基因组的序列id是不是对应,搜索检查一下;
5 老师,我在学习基因家族视频,用perl脚本get_gene_weizhi预测基因顺式作用元件时出现了问题,老师请看
回答问题即可获得 10 经验值,回答被采纳后即可获得 10 金币。
老师,是这样的,我的物种是川桑(Morus notabilis),没有收录在在Ensembl数据库,所以我是在川桑数据库下载的genome,gene,cds及protein。如下图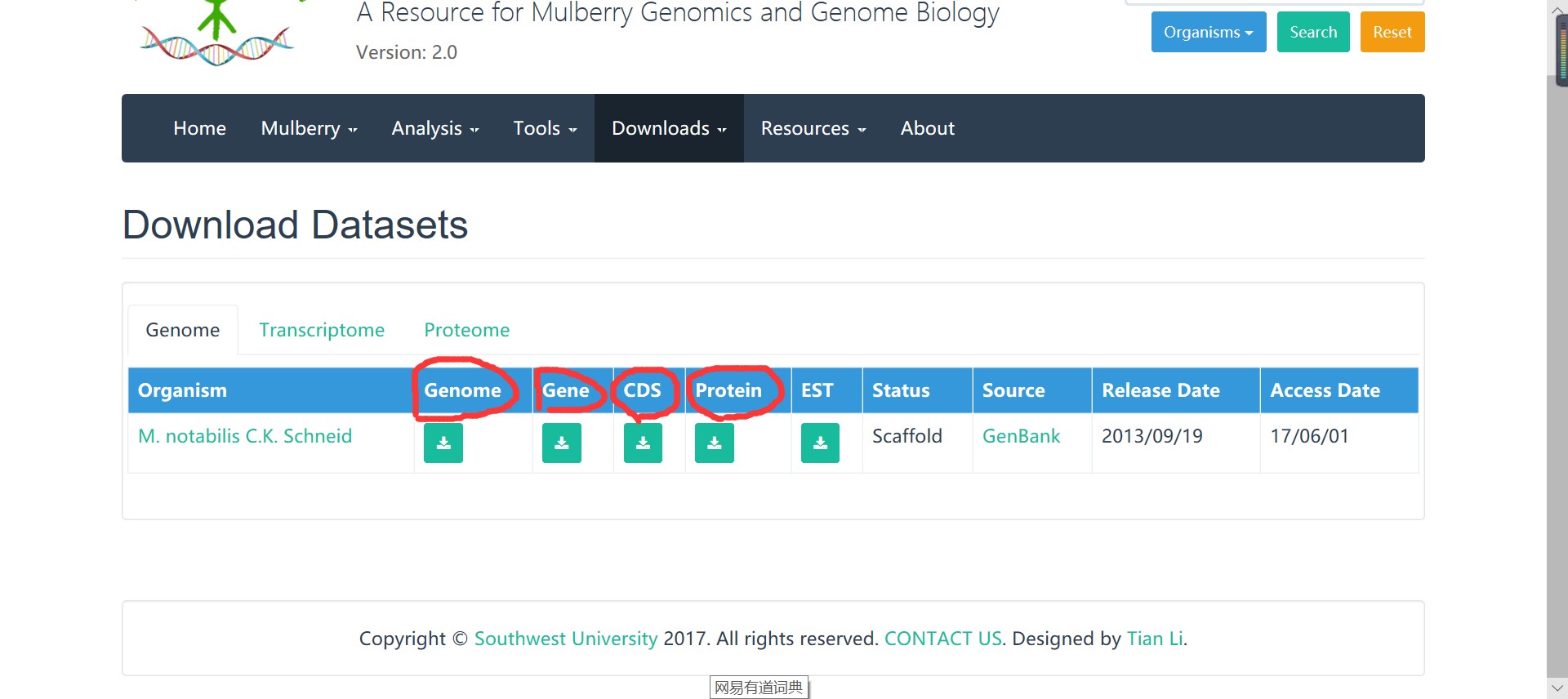
然后在NCBI上下载了川桑的GFF注释文件,如下图
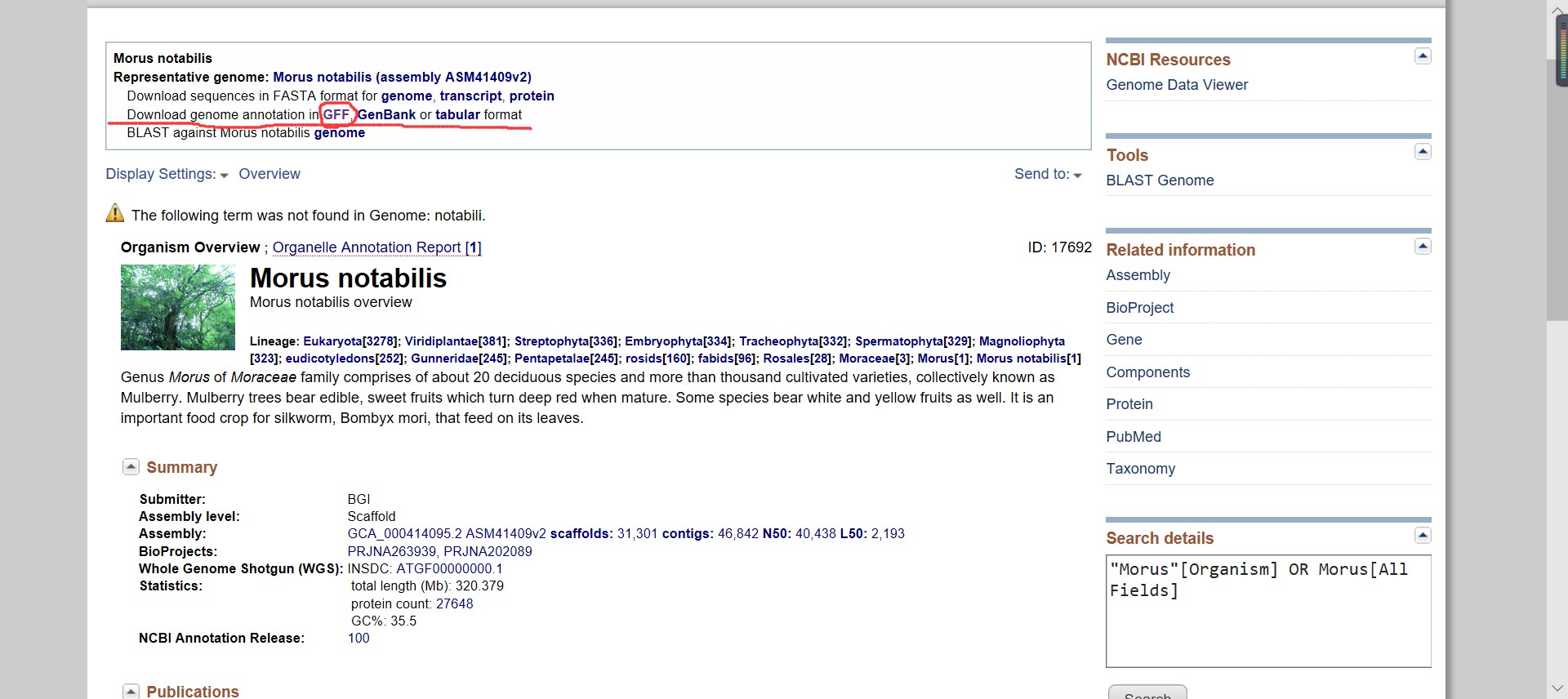
老师,下图是我根据视频学习找到的基因ID 然后运行perl脚本get_gene_weizhi,得到gene_weizhi.txt文件,如下图
然后运行perl脚本get_gene_weizhi,得到gene_weizhi.txt文件,如下图
老师,这儿我补充一点,川桑基因组数据库没有基因在哪条染色体上的信息,所以这里的结果就没有第二列出现基因ID的染色体位置,图中应该是基因在某个片段上的信息,我猜想不管是在染色体上还是在染色体片段上,应该都能找到上游1500位置的序列,所以我就接着往下走,运行Perl脚本tiquxulie,这里perl脚本后面跟的是genome,然后界面上就会出现如下信息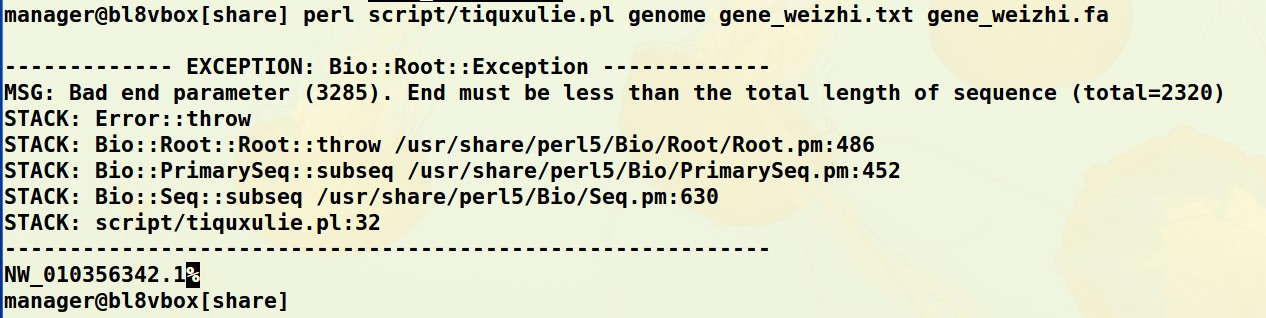
然后查看gene_weizhi.fa,得到如下结构,只有一列短的波浪线
然后我又运行这个脚本,只是perl脚本后面跟的是morus_notabilis.gene.fa,然后出现提示信息
然后查看Ggene_weizhi.fa,跟上面一样的结果
老师,我这个是怎么回事儿呀,我又是初入这个领域,才学疏浅,望老师指导!
2 个回答

老师,我的基因位置信息里面的ID和基因组的ID是一样的。
可能我没有把问题说清楚,我在运行perl脚本,get_gene_weizhi.pl后得到的基因位置信息是这样的
下图是GFF文件的一部分

get_gene_weizhi.pl得到了一共2万多个结果,我的基因一共才六十几个,我查看了一下GFF文件,发现这2万多个结果(就是NW开头这一栏)是GFF里面所有注释的gene,也就是说不是按照我的基因ID来提取的基因位置,而是将所有基因位置都提取了出来。并且第一幅图里面,得到的位置信息没有第一列(也就是基因ID),第二列是全基因组鸟枪序列(因为我们的这个物种没有基因在染色体位置的信息),第三列跟第四列都正常,分别是基因的位置和正负链,所以,我猜想,因为我的基因位置文件缺了一列,所以最后提取不了基因上游的1500序列,可是,老师,我运行get_gene_weizhi.pl后,为什么把GFF里面所有的gene都提取了出来呀,还有第一列的基因ID怎么不见了。下面是我的基因组数据的一部分。NW是鸟枪序列。
老师,我这个是怎么回事儿呀