不好意思,第一次提问,下次一定写的详细来问您
我是这样运行的:
先创建了pep.fa文件
然后执行
[share] perl get_fa_by_id.pl AT.gff TAIR10.pep.all.fa pep.fa
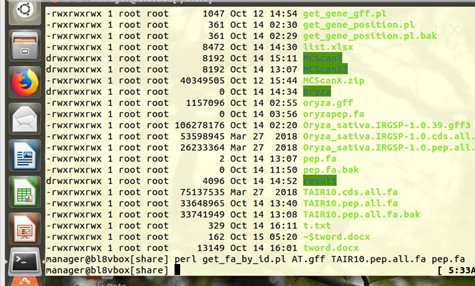
AT.gff是之前提取的基因位置信息,没有问题
运行完没有报错,但
pep.fa文件是0字节
不好意思,第一次提问,下次一定写的详细来问您
我是这样运行的:
先创建了pep.fa文件
然后执行
[share] perl get_fa_by_id.pl AT.gff TAIR10.pep.all.fa pep.fa
AT.gff是之前提取的基因位置信息,没有问题
运行完没有报错,但
pep.fa文件是0字节
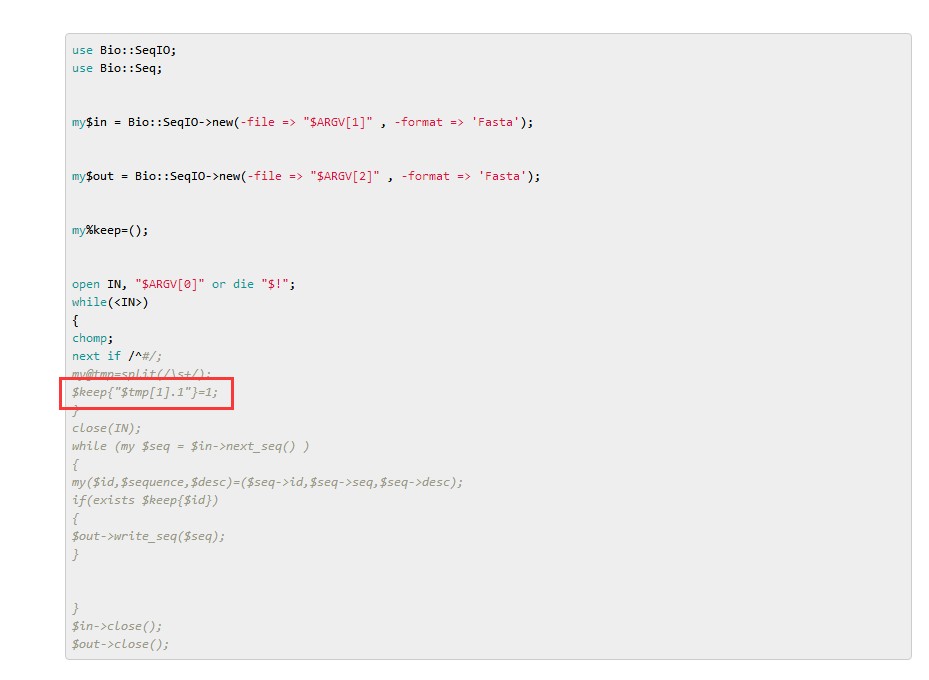
get_fa_by_id.pl 这个脚本是根据第一个文件信息的第一列ID信息,提取第二个文件fasta文件的中对应的序列信息,存储到第三个文件当中;所有两个文件当中的ID对应才能得到想要的序列;
分析代码:
代码中这个地方 给ID都加了个".1" 你看看你的gff 文件里面的第一列ID都加 .1 后的基因ID,与你提供的所有蛋白质序列里面的序列ID能不能对应?如果不能对应请改正。
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
