5 超大的文本文件如fasta,fastq文件在windows中如何打开
最佳答案 2018-07-22 07:00
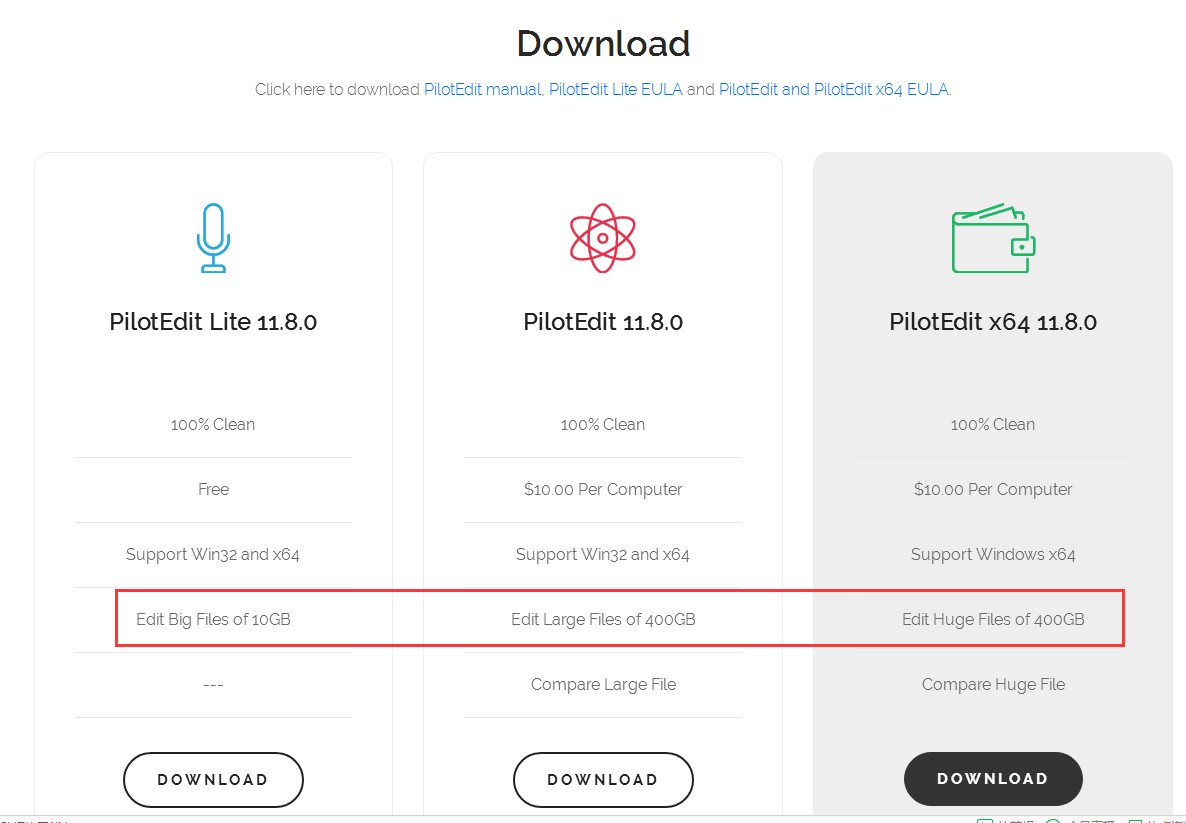
这里推荐pilotedit软件(下载地址:http://www.pilotedit.com/index.html),专门用于windows当中打开超大文本文件,免费版本就可以打开最大10G的文件;收费版本甚至400G的文件都么有问题;
如果在linux当中就简单多了,用less命令就可以。
更多fastq说明《illumina二代测序原理及fastq视频课程》
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读、
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

其它 1 个回答
其实gedit可以打开fastq文件,只是因为3G文件太大gedit就有点力不从心了,可以写个小程序跑截取部分,这样文件变小就可以用gedit打开,
c语言:
#pragma warning(disable:4996)
#include <stdio.h>
#include <iostream>
int main()
{
int m;
char ch;
FILE *fqout, *fqin;//原文件指针,新文件指针
fqout = fopen("E:\\Administrator\\Source\\repos\\SRR539135_2.fastq","r+");
fqin = fopen("E:\\Administrator\\Source\\repos\\SRR539135_2_copy.fastq","w+");
if (fqout == NULL)
{
perror("open file_out failed!\n");//打开失败提醒
}
if (fqin == NULL)
{
perror("open file_in failed!\n");
}
printf("please input the number of copying lines :\n");
scanf("%d",&m);
m = 4 * m;//复制m行到新文件中,之所以乘4,因为fastq是每4行在一起的,
while (m)
{
while ((ch = fgetc(fqout)) != '\x0a')//复制一行到新文件中,
{
fputc(ch, fqin);
}
fputc(ch, fqin);
m--;
}
fclose(fqin);
fclose(fqout);
return 0;
}
程序粗糙,只是想帮忙,不好意思~~