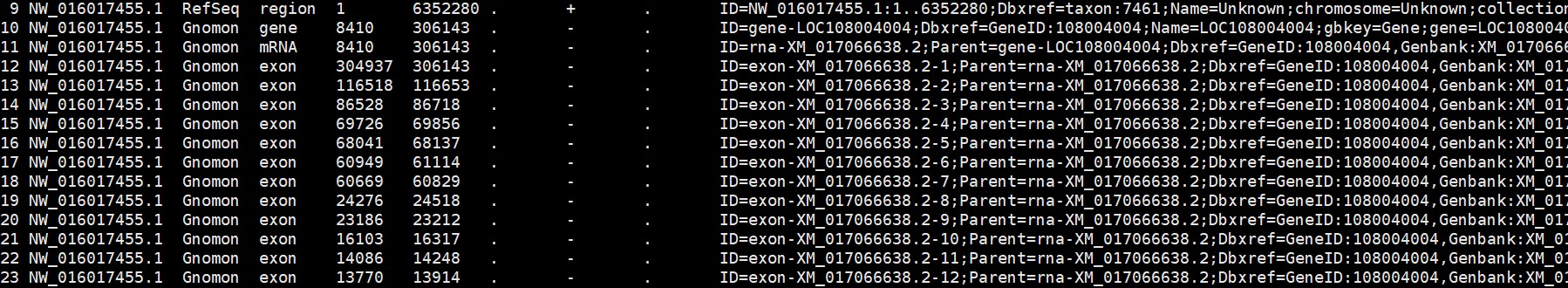
老师,您好,我在学习您的课程(RNAseq自主分析)中,使用docker分析转录组获取全长时候总是出现报错,命令行为,python ../scripts/get_gene_length_from_gtf.py -g genome.gtf -p gene_length;报错信息为:
GTF 文件不标准,有些行缺少gene_id
可能是非编码的基因,可以删除这些行,再重新运行代码:
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
老师,您好,我看了一下gff文件,里面有gene注释信息,好像和文章中的问题不一样,您能再看下吗,谢谢!
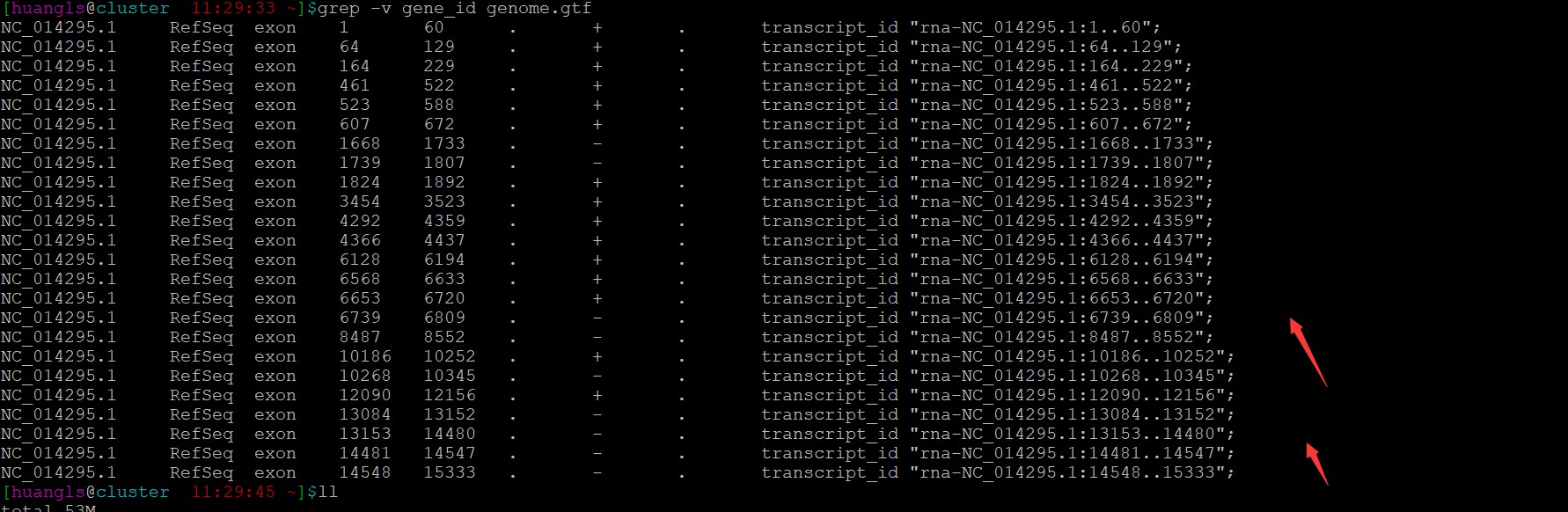
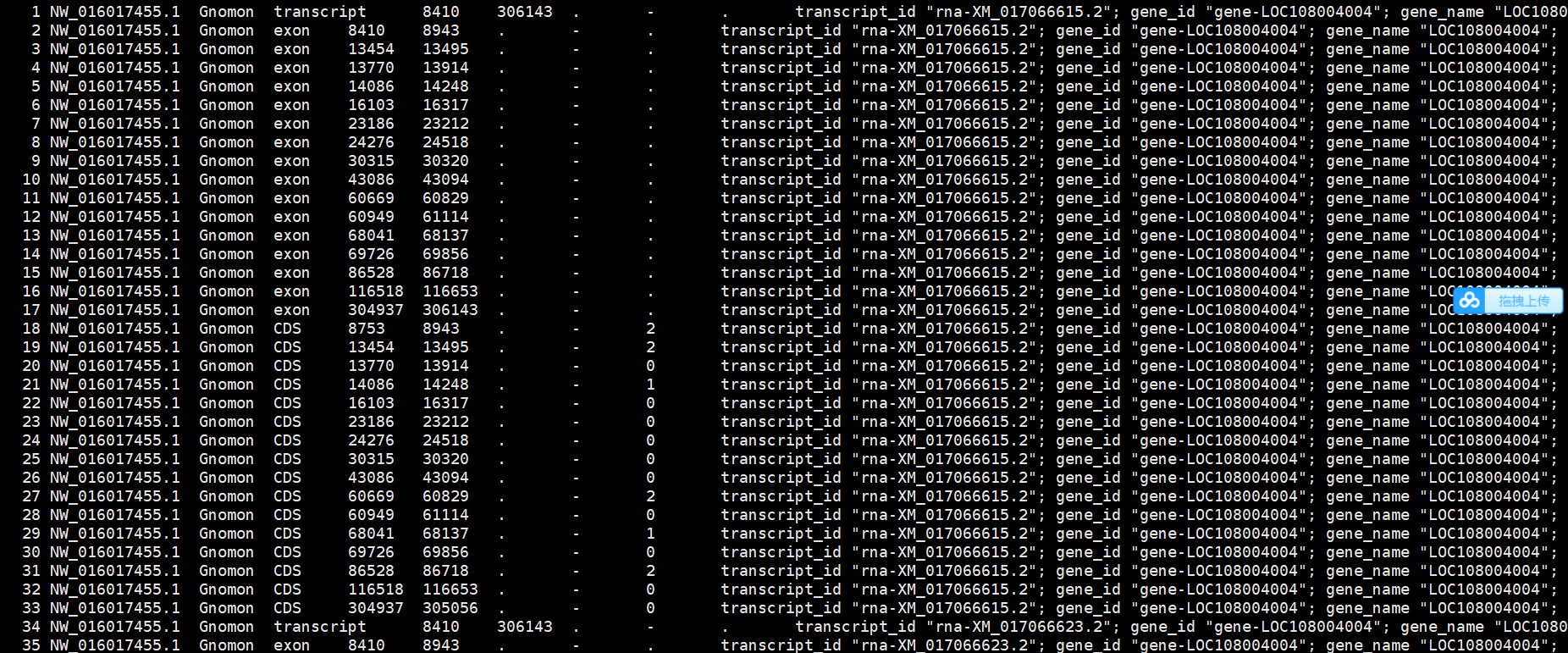 请教老师如何解决这个问题,不胜感激!
请教老师如何解决这个问题,不胜感激!