你好,我也遇到了同样的问题,请问你解决了吗?
蛋白三维结构预测遇到问题:当在swiss-model中选择使用模板的方法时,在上传模板的时候发现因为以下问题没法上传
这个就是蛋白序列文件和其经过比对得到的模板文件protein sequences.txt 4mla.pdb。
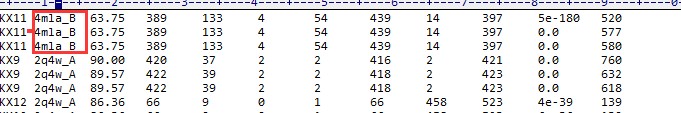
利用黄老师讲的方法,从PDB数据库中下载经过PSI-BLAST比对获得的模板的PDB格式文件,在swiss-model中,使用自己模板的方法进行三维结构预测,结果在上传文件的时候遇到以上的问题,请问怎么解决。
Chains A, B of the uploaded template do not start with residue no. 1. Will be renumbered.
Chain A of the uploaded template contains multiple unordered residues, starting at residue 623. Residue numbers must occur in ascending order. That way, the structure does not apply for modelling
个人感觉是PDB文件是不是出现不标准,残基的排序有问题,需要重新排序,这样的话,应该怎么弄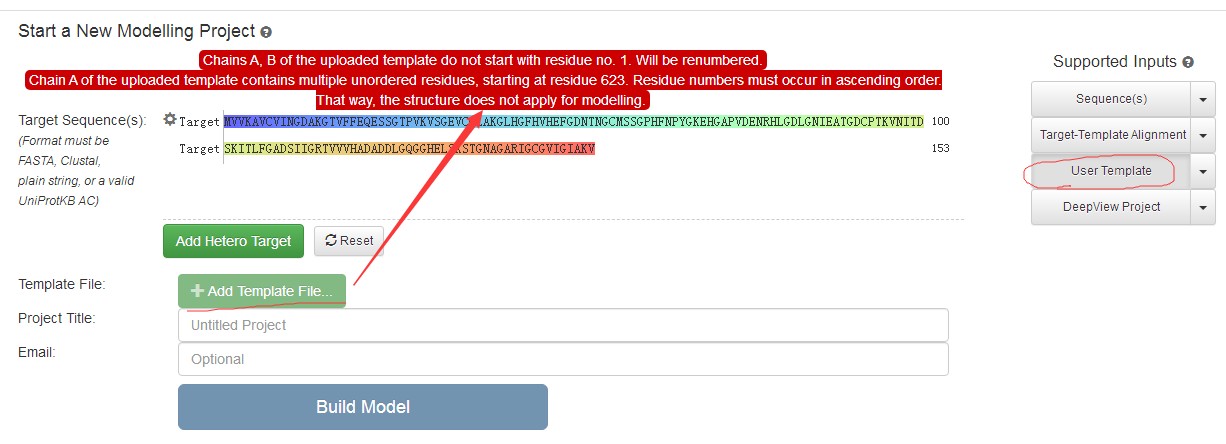
Chains A, B of the uploaded template do not start with residue no. 1. Will be renumbered.
Chain A of the uploaded template contains multiple unordered residues, starting at residue 623. Residue numbers must occur in ascending order. That way, the structure does not apply for modelling
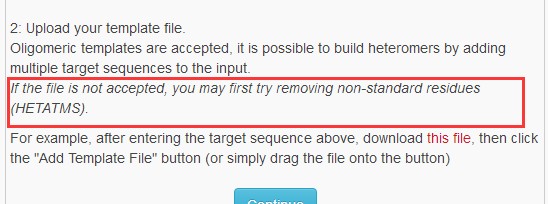
另外就是我在旁边的帮助一栏中发现如下,是不是这个的原因?
 补充:我又利用黄老师给的Python的程序下载这个蛋白模板的PDB格式,结果下载的是CIF格式的该文件4mla.cif
补充:我又利用黄老师给的Python的程序下载这个蛋白模板的PDB格式,结果下载的是CIF格式的该文件4mla.cif
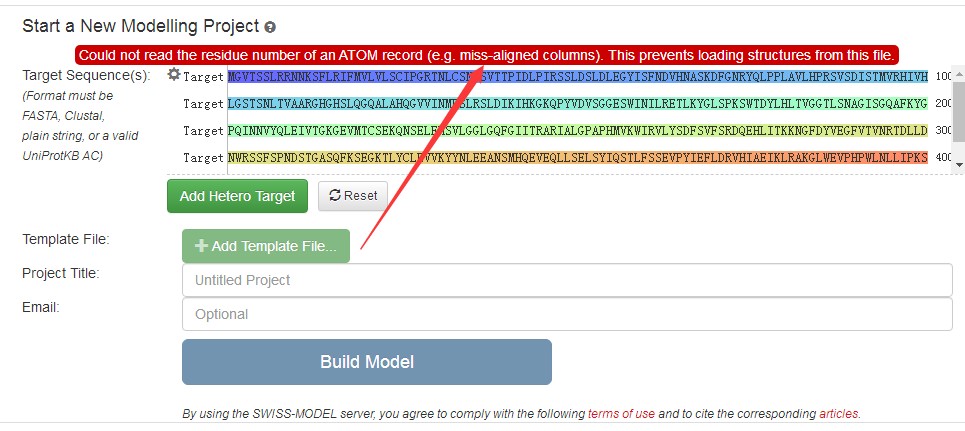
然后将这个文件上传为模板时,又出现如下的问题:
Could not read the residue number of an ATOM record (e.g. miss-aligned columns). This prevents loading structures from this file