10 利用samtools将sam文件转化为bam文件时,sam文件报错
运行命令:
$samtools sort -@ 4 -O bam -o RNA.bam RNA.sam
报错:
[E::sam_hdr_create] Invalid header line: must start with @HD/@SQ/@RG/@PG/@CO
samtools sort: failed to read header from "RNA.sam"
RNA.sam文件行首:
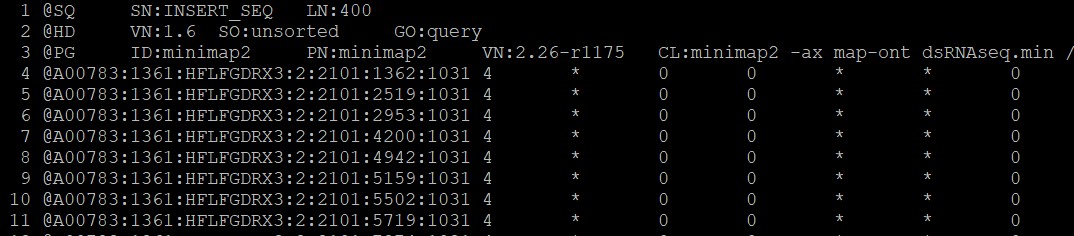
最佳答案 2023-10-13 10:44
是在上一步生成sam文件时出现问题。
命令:
minimap2 -ax map-ont RNAseq.min RNA.clean.fa.gz >RNA.sam
在这一步中使用的测序结果数据RNA.clean.fa.gz是fasta格式的,错误。应该是fastq格式的才可以