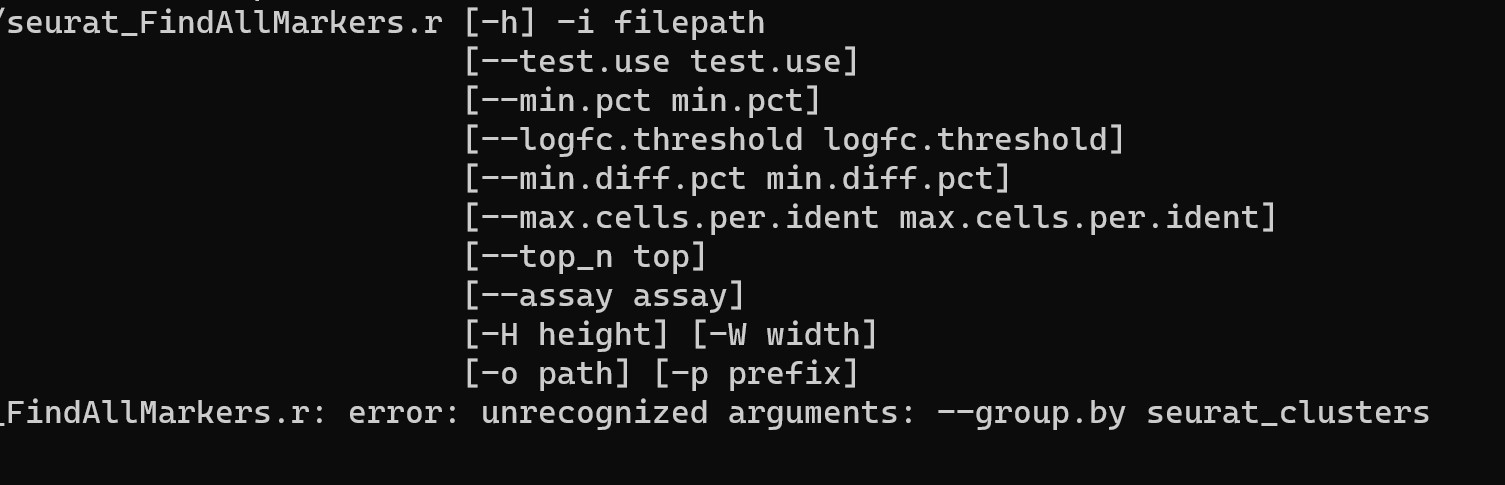
老师好!我想在注释细胞亚群之前提取不同Cluster的Marker基因,但课程讲解和提供的代码是先注释不同亚群再进行不同亚群Marker基因的提取,代码如下:Rscript $scripts/seurat_FindAllMarkers.r --rds $workdir/05.cell_type_ann/pbmc.added.celltype.rds \
-p FindAllMarkers --test.use DESeq2 --logfc.threshold 0.25 。我执行后报错:Warning: When testing 14 versus all:
every gene contains at least one zero, cannot compute log geometric means
data frame with 0 columns and 0 rows
Error in `group_by_prepare()`:
! Must group by variables found in `.data`.
• Column `cluster` is not found.
Backtrace:
▆
1. ├─obj.markers %>% group_by(cluster) %>% ...
2. ├─dplyr::top_n(., n = opt$top_n, wt = avg_log2FC)
3. │ └─dplyr::filter(...)
4. ├─dplyr::group_by(., cluster)
5. └─dplyr:::group_by.data.frame(., cluster)
6. └─dplyr::group_by_prepare(.data, ..., .add = .add, caller_env = caller_env())
7. └─rlang::abort(c("Must group by variables found in `.data`.", glue("Column `{unknown}` is not found.")))
Execution halted 想请教老师应该怎么解决?谢谢老师