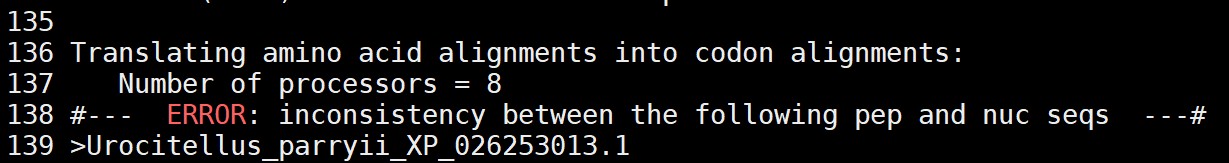
老师好,我写了sh后台运行ParaAT 多序列比对查看运行完成的日志文件时,大多数物种都出现这个报错情况:
在将氨基酸序列(pep)转换为核酸序列(nuc)时发现了不一致性
并且继续用这个结果进行下一步的合并分析时#合并成supergene
seqkit concat ./aln_out/*.pep_aln.renamed > single_copy.pep_msa.fasta
输出的结果是空的……
前面都是按照课程的脚本跑的
请问该怎么处理这个报错呢?
这里有基因ID,你找到对应ID的cds和蛋白序列检查他们之间是不是3倍关系;确认解决一下;
如果数量不多,可以把这样的基因对删除
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!