同学,你执行R脚本的时候-c 参数应该给的是分析的染色体编号,我看你给的这个染色体编号怎么好像和gff文件里对不上,你再检查一下看看是不是,另外再提醒你一下,这个-s 和-e的参数是你要分析的所有snp位点的最开始那个snp和最后一个snp位点的位置(如果是基因的话就是基因在染色体上的起始和终止位置),不要填错了哈
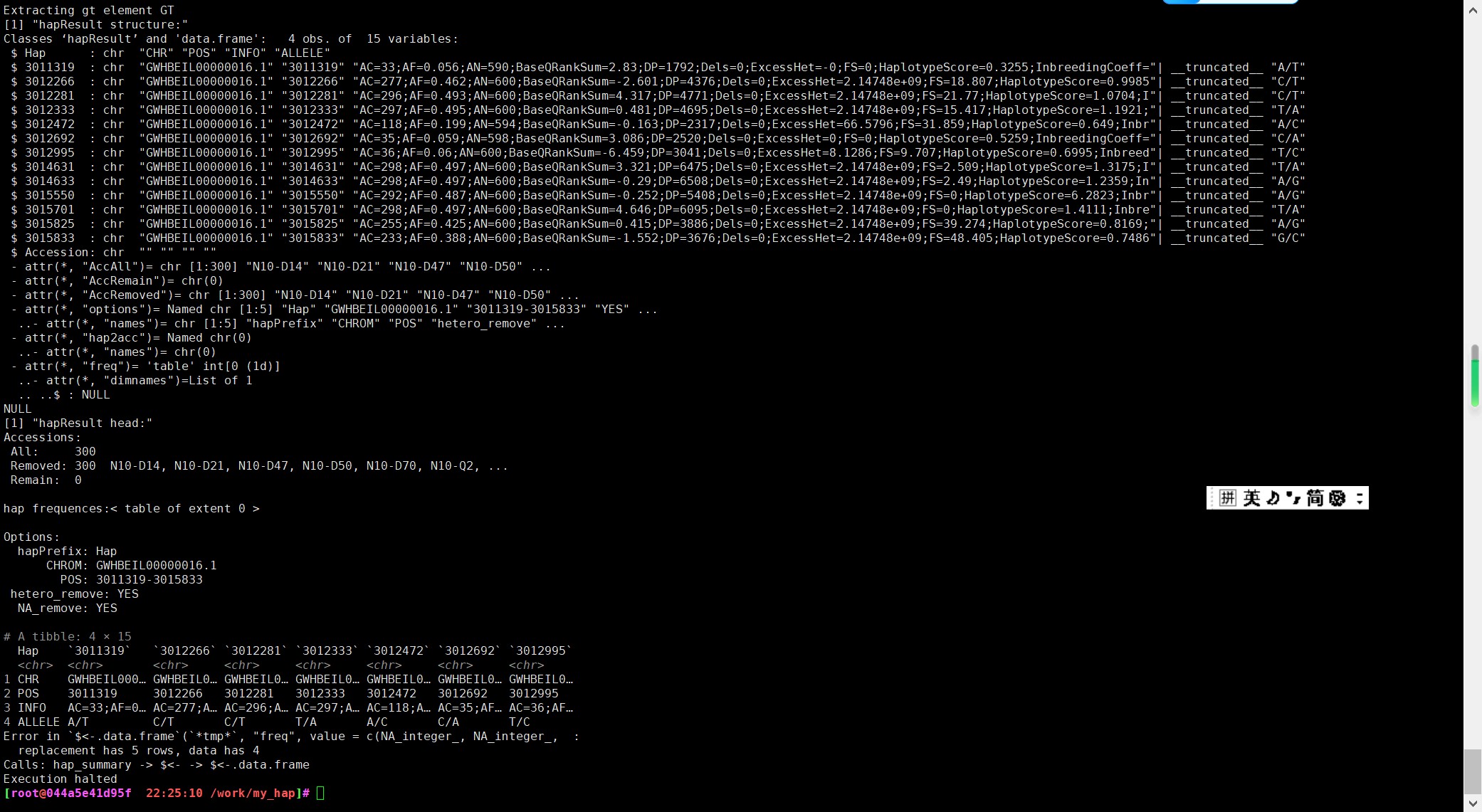
50 单倍型报错
回答问题即可获得 10 经验值,回答被采纳后即可获得 55 金币。
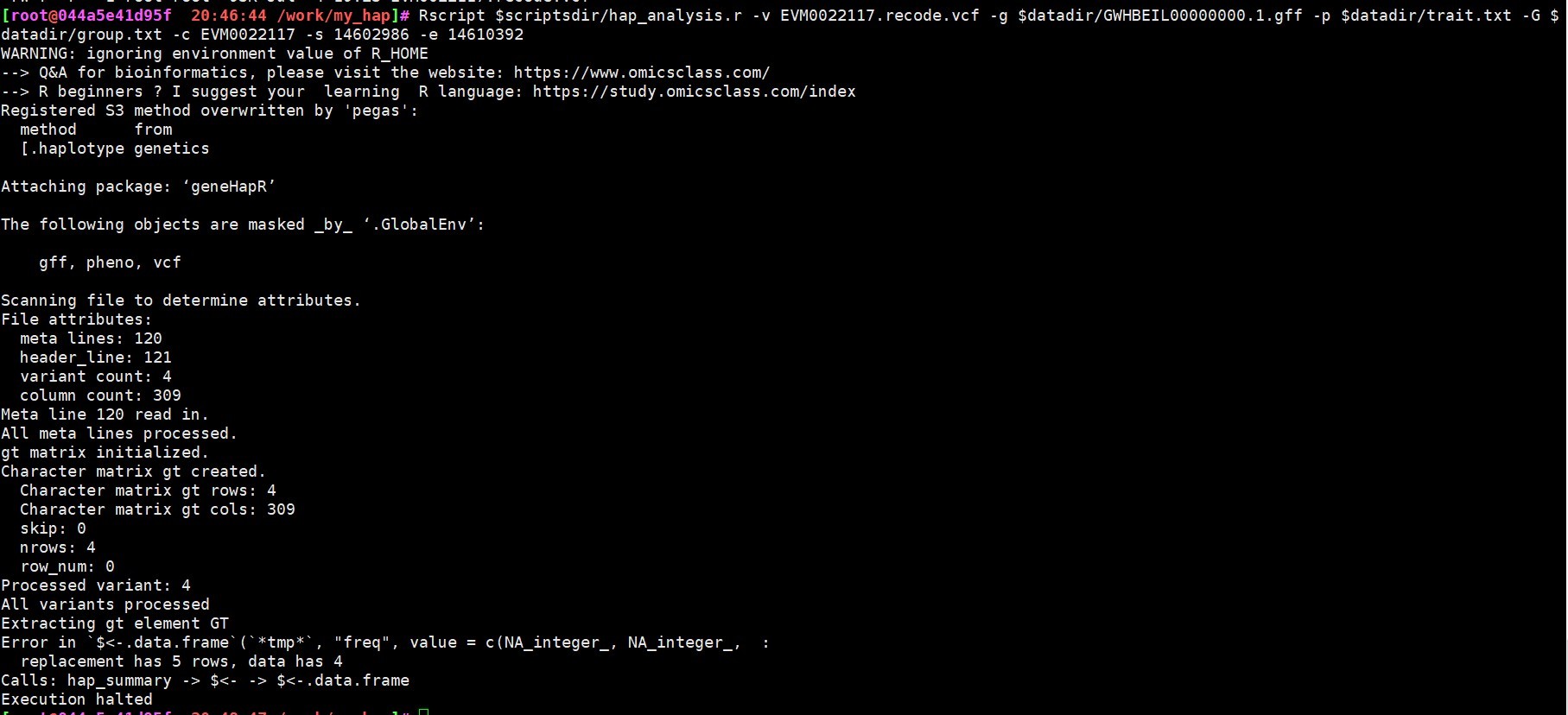 报错信息
报错信息
 分组格式
分组格式
 gff格式
gff格式
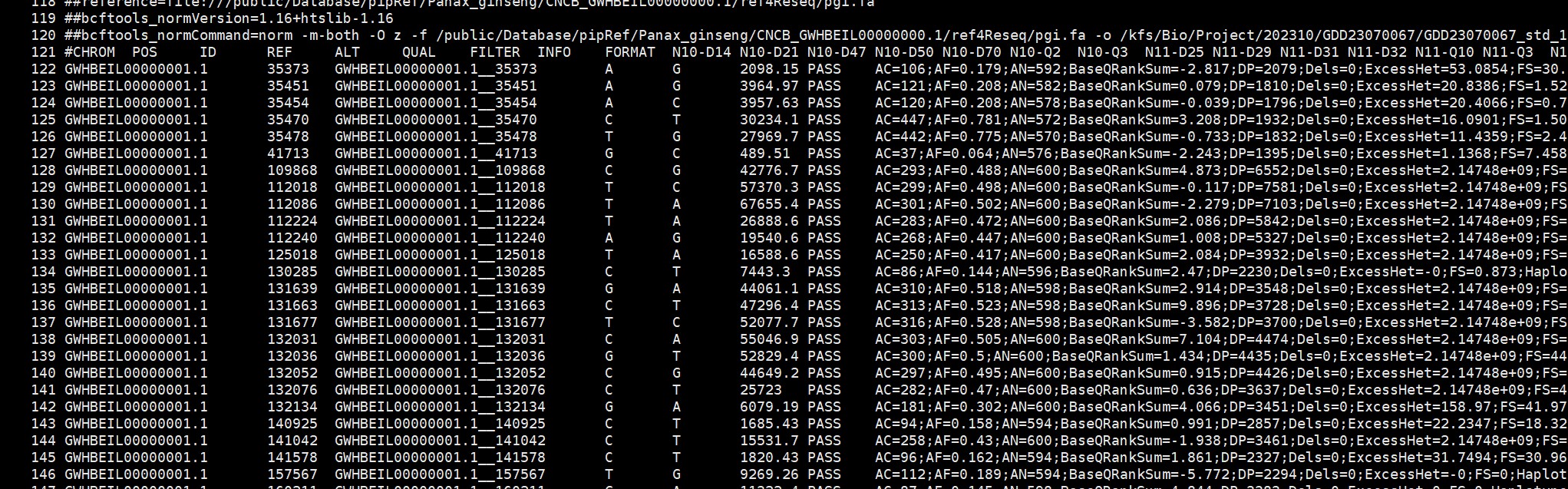 vcf格式
vcf格式
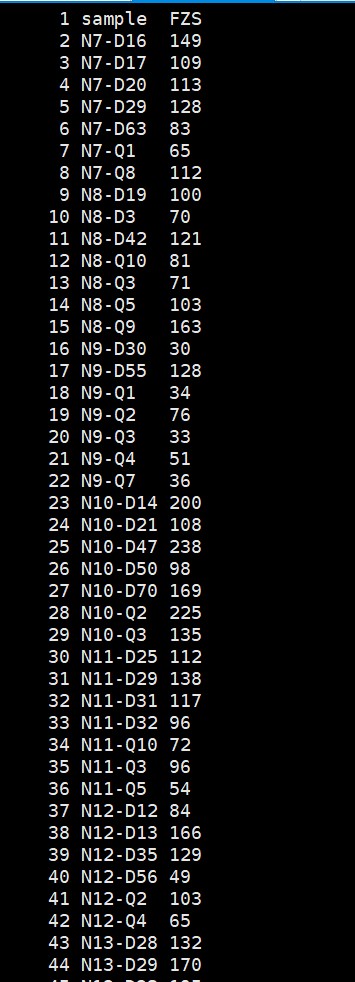 表型格式
表型格式
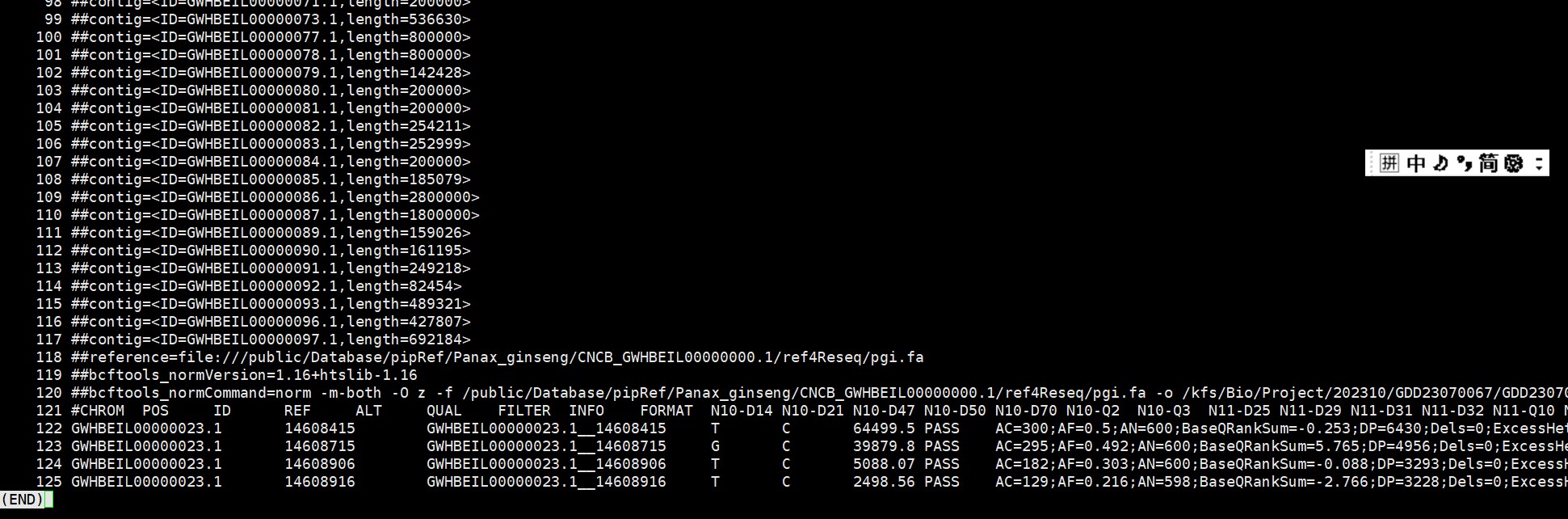 EVM0022117.recode.vcf
EVM0022117.recode.vcf
脚本如下:
library(argparse) # 导入包
# Add command line arguments
parser <- ArgumentParser(description='haplotype analysis plot')
parser$add_argument( "-i", "--workdir", type="character",default=getwd(),
help="input file directory [default %(default)s]",
metavar="filepath")
parser$add_argument( "-v", "--vcf", type="character",required=T,
help=" vcf files[required]")
parser$add_argument( "-g", "--gff", type="character",required=T,
help=" gff files[required]")
parser$add_argument( "-p", "--pheno", type="character",required=T,
help=" pheno files[required]")
parser$add_argument( "-G", "--group", type="character",required=T,
help=" group files[required]")
parser$add_argument( "-c", "--chr", type="character",required=T,
help=" chr name[required]")
parser$add_argument( "-s", "--start", type="double",required=T,
help=" start position[required]",
metavar="number")
parser$add_argument( "-e", "--end", type="double",required=T,
help=" end position[required]",
metavar="number")
parser$add_argument( "-o", "--outdir", type="character", default=getwd(),
help="output file directory [default %(default)s]",
metavar="path")
opt <- parser$parse_args()
if( !file.exists(opt$outdir) ){
if( !dir.create(opt$outdir, showWarnings = FALSE, recursive = TRUE) ){
stop(paste("dir.create failed: outdir=",opt$outdir,sep=""))
}
}
###############################################################################
####### 一些基本参数的设置
vcf <- opt$vcf # 感兴趣位点的vcf文件
gff <- opt$gff # gff文件
pheno <- opt$pheno # 表型数据
group <- opt$group # 样本分组信息
Chr <- opt$chr # 基因所处的染色体名称
start <- opt$start # 基因的起始位置(染色体坐标)
end <- opt$end # 基因的终止位置(染色体坐标)
hapPrefix <- "Hap" # 单倍型名称的前缀
# 安装及加载包
#install.packages("geneHapR")
library(ggplot2)
library(geneHapR)
# 设置工作目录
setwd(opt$workdir)
####### 1.从数据导入到单倍型鉴定
# 导入各种数据
vcf <- import_vcf(vcf) # 导入VCF格式的基因型数据,建议先提取感兴趣的位点
gff <- import_gff(gff) # 导入GFF格式的注释数据
pheno <- import_AccINFO(pheno) # 导入表型数据
group <- import_AccINFO(group, header=T,
sep = "\t", # 分隔符号,默认为制表符"\t"
na.strings = "NA") # 导入其他样本信息
# 从VCF开始单倍型鉴定
hapResult <- vcf2hap(vcf, hapPrefix = hapPrefix,
hetero_remove = TRUE, # 移除包含杂合位点的样本
na_drop = TRUE) # 移除包含基因型缺失的样本
# 对单倍型结果进行汇总整理
hapSummary <- hap_summary(hapResult, hapPrefix = hapPrefix)
# 将单倍型鉴定结果导出
write.hap(hapResult, file = "hapResult.txt")
write.hap(hapSummary, file = "hapSummary.txt")
##### 2.单倍型结果可视化分析
# 以表格形式展示各单倍型的基因型
pdf("HapTable.pdf",width=8,height=6)
plotHapTable(hapSummary, # 单倍型结果
hapPrefix = hapPrefix, # 单倍型名称前缀
angle = 45, # 物理位置的角度
displayIndelSize = 0, # 图中展示最大的Indel大小
title = "HapTable") # 图片标题
dev.off()
#### 3.单倍型网络分析
hapSummary[hapSummary == "DEL"] = "N"
hapnet <- get_hapNet(hapSummary, # 单倍型结果
AccINFO = group, # 包含样本分类信息的数据框(data.frame)
groupName = "group", # 含有样本分类信息的列名称
na.label = "Unknown") # 未知分类样本的类别
pdf("HapNet.pdf",width=8,height=6)
plotHapNet(hapnet, # 单倍型网络
scale = "log2", # 标准化方法"log10"或"log2"或"none"
show.mutation = 2, # 是否展示变异位点数量, 0,1,2,3
col.link = 2, link.width = 2, # 单倍型之间连线的颜色和宽度
main = "HapNet", # 主标题
pie.lim = c(0.5, 2), # 圆圈的大小
legend_version = 1, # 图例形式(0或1)
labels = T, # 是否在单倍型上添加label
legend = c(5,5), # 图例的坐标
cex.legend = 0.8) # 图例中文字的大小
dev.off()
##### 4.连锁不平衡分析
pdf("LDheatmap.pdf",width=8,height=6)
plot_LDheatmap(hap = hapResult, # 单倍型结果
add.map = TRUE, # 是否添加基因模式图
gff = gff, # 注释信息
Chr = Chr, # 染色体名称
start = start, # 基因的起始位置
end = end) # 基因的终止位置(更多参数参见帮助文档)
dev.off()
###### 5.表型关联分析
# 单个表型的分析
pdf("firstPheno.pdf",width=8,height=6)
hapVsPheno(hap = hapResult, # 单倍型分析结果
pheno = pheno, # 表型
hapPrefix = hapPrefix, # 单倍型名称的前缀
title = "hapVsPheno", # 主标题
minAcc = 4, # 参与p值计算所需的最小样本数
symnum.args = list( # 定义显著性标注方式
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")),
mergeFigs = FALSE) # 结果包括两个图,是否融合成一张图
dev.off()
# 多个表型的分析
hapVsPhenos(hap = hapResult,
pheno = pheno,
hapPrefix = hapPrefix,
title = "hapVsPhenos",
compression = "lzw", # tiff文件的压缩方式
res = 300, width = 16, height = 8, # 图片大小的单位"inch"
outPutSingleFile = TRUE, # 只有pdf格式支持输出单个文件
filename.surfix = "pdf", # 文件格式: pdf, png, jpg, tiff, bmp
filename.prefix = "hapVsPhenos") # 文件名称为: prefix + pheno_name + surfix
# 逐位点表型显著性分析
pdf("PhenoPerSite.pdf")
hapVsPhenoPerSite(hap = hapResult, # 单倍型分析结果
pheno = pheno, # 表型文件
phenoName = "heading_days", # 表型名称
freq.min = 5) # 参与显著性计算的最小样本数
dev.off()
# 回车继续下一位点
# ESC退出当前命令