单倍型分析报错
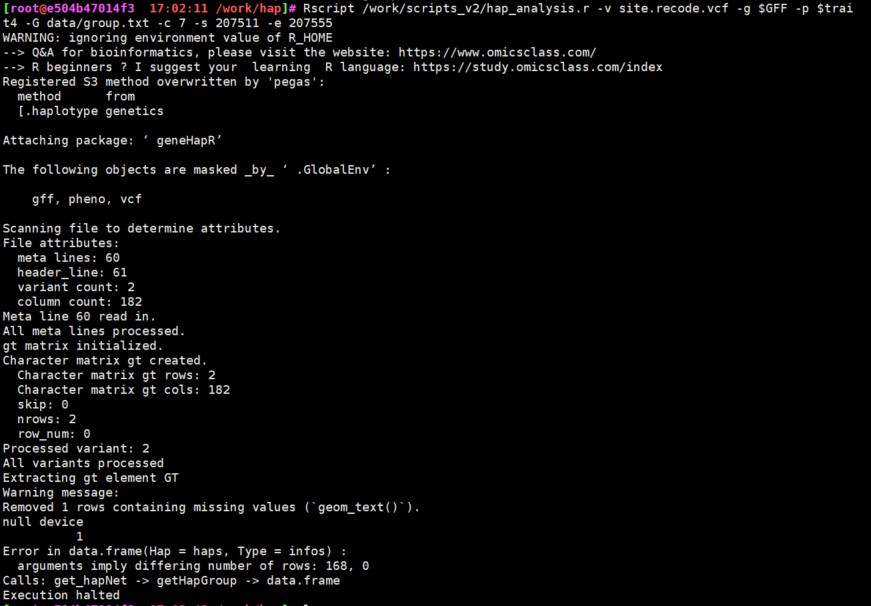
我的问题是这样的,在进行单倍型分析的时候,利用教程给的demo数据,按照w.sh中的代码一步步操作,能正常分析,非常丝滑,但是我只要换成自己的数据,就会报错“Error in data.frame(Hap = haps, Type = infos) : arguments imply differing number of rows: 168, 0”。我已经检查了自己的文件,包括gff3文件,site文件,表型文件以及分组文件,对比了demo文件,可以非常确定,这些文件没有任何问题。进一步发现,在我的群体(共173个基因型)中,确实有5个基因型在目标size上是杂合或存在缺失,但是我查看了demo数据也发现,demo使用的群体是176个基因型,其中只有113个基因型在site位置是纯合的,但是它却没有因此而报错。另外,导致深究了以上报错的原因,发现是Error: hapSummary 和 group 文件的行数不一致。但是这两个文件本身完全不同,正常情况下就应该行数不一致。最后,我尝试了社区之前有人提供的解决方案,即将hap_analysis.r 脚本中hetero_remove = TRUE和na_drop = TRUE改成:hetero_remove = FALSE,na_drop = FALSE,但结果还是会报错。
报错界面如下:
运行的命令如下:
Rscript /work/scripts_v2/hap_analysis.r -v site.recode.vcf -g $GFF -p $trait4 -G data/group_delete.txt -c 7 -s 207511 -e 207555
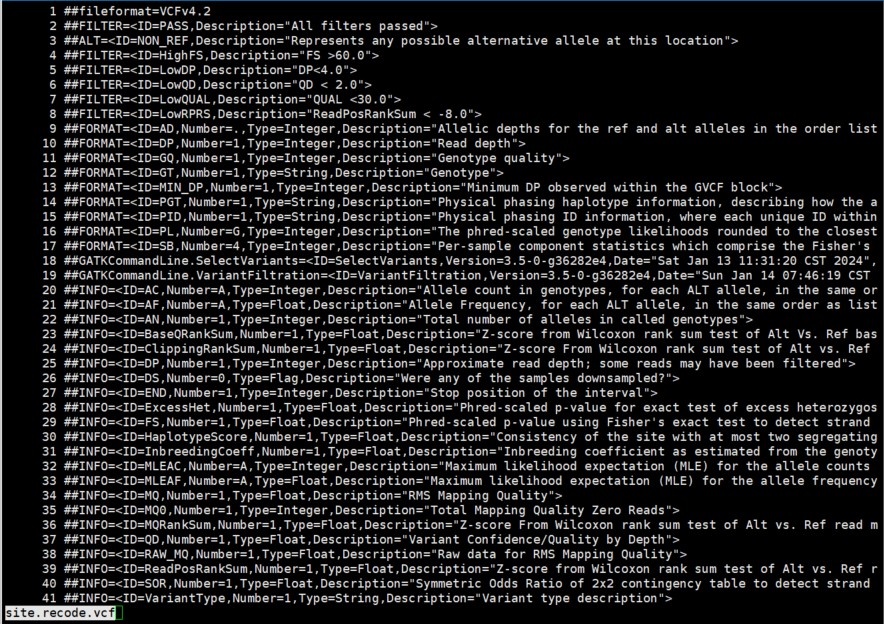
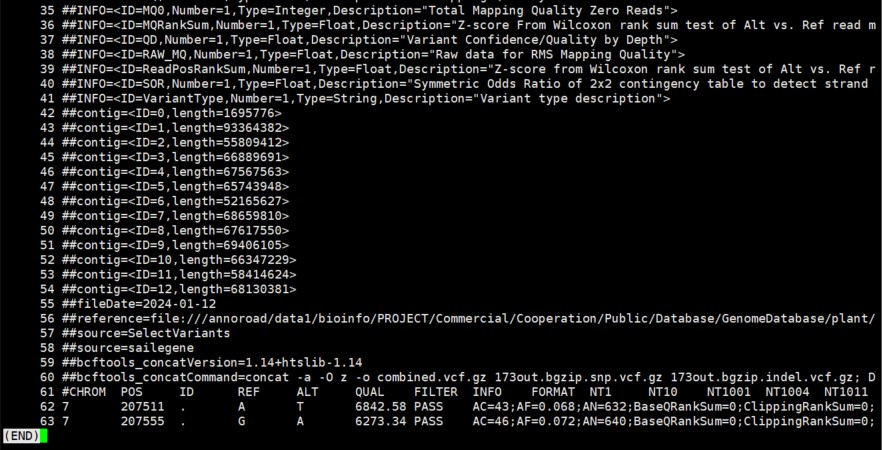
其中的site.recode.vcf文件:
其中的$GFF:
 其中的$trait4如下:
其中的$trait4如下:
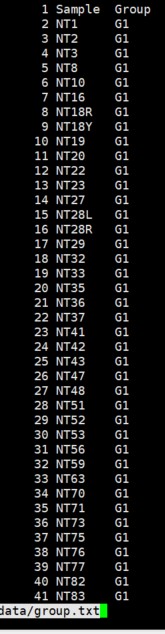
 其中的data/group.txt :
其中的data/group.txt :