建议用命令检查染色体数量:
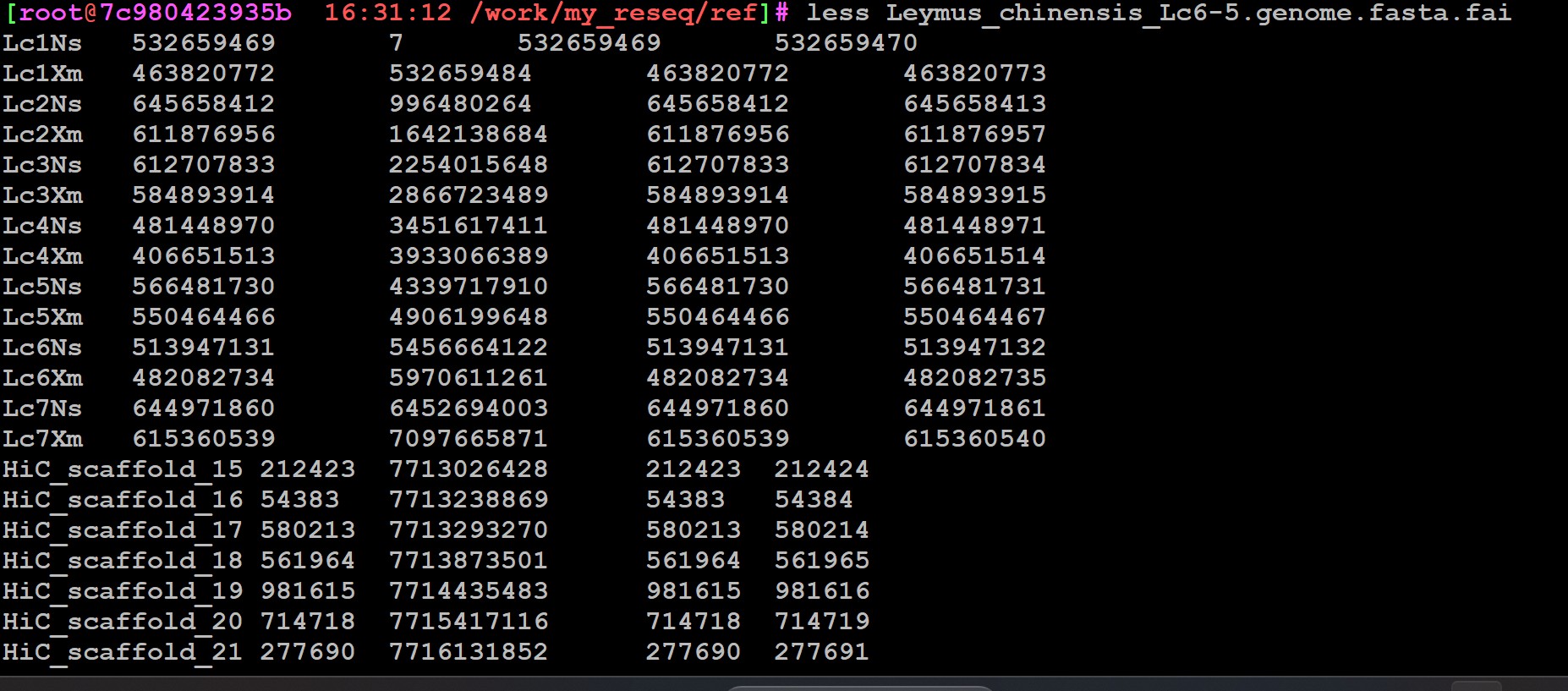
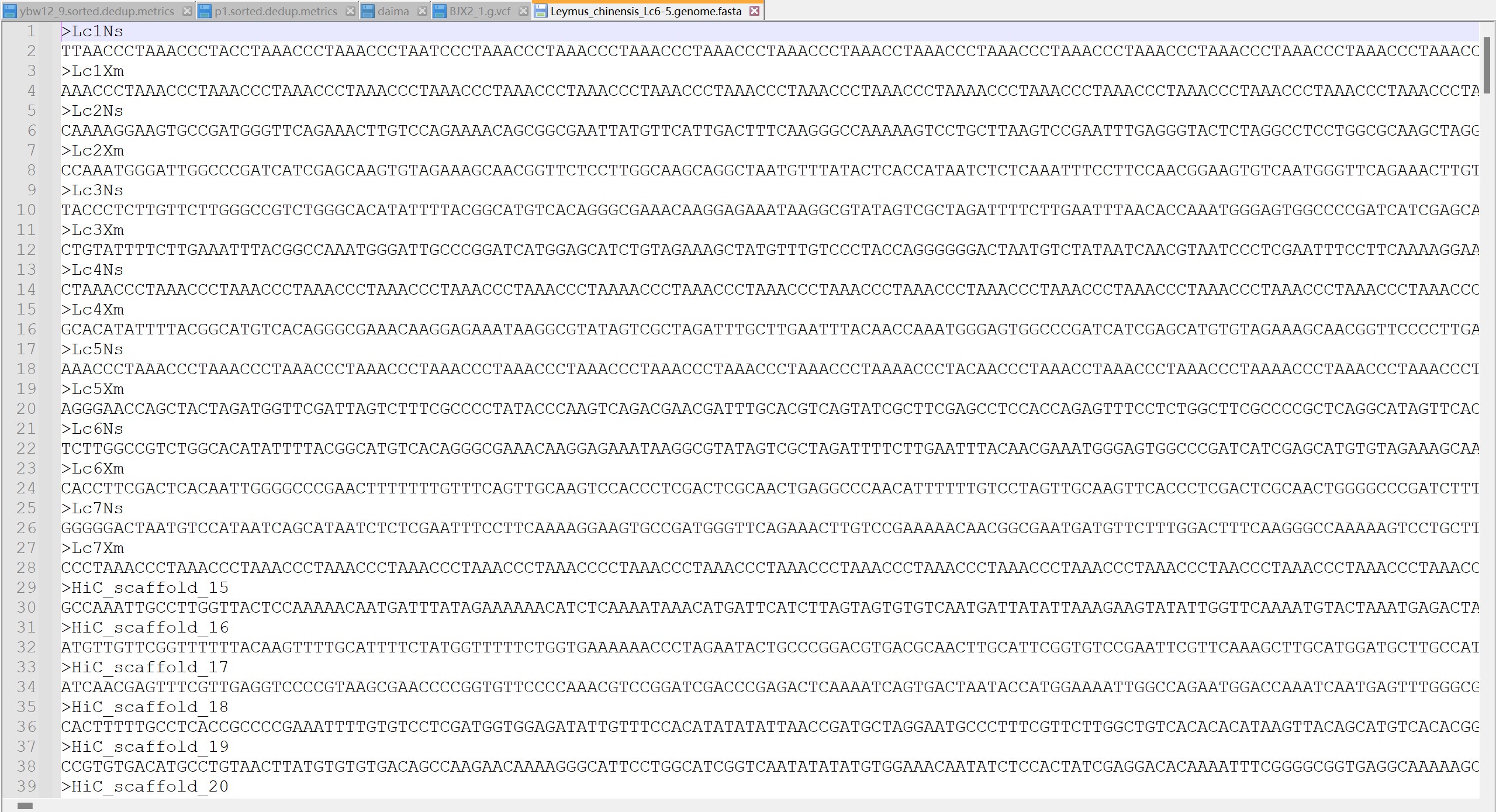
cut -f 1 demo.vcf| sort|uniq -c #压缩的zcat demo.vcf.gz|cut -f 1 | sort|uniq -c
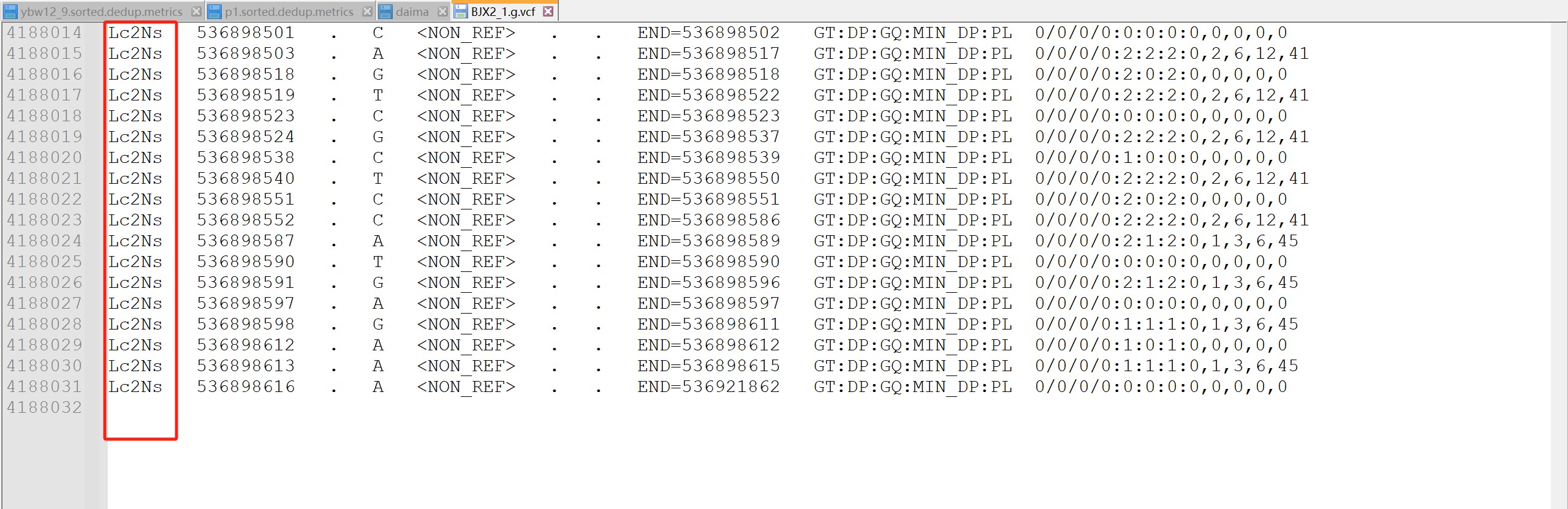
检查gvcf 里面的数据是不是全的。不全重新跑,输出的时候输入文件后缀不要加gz
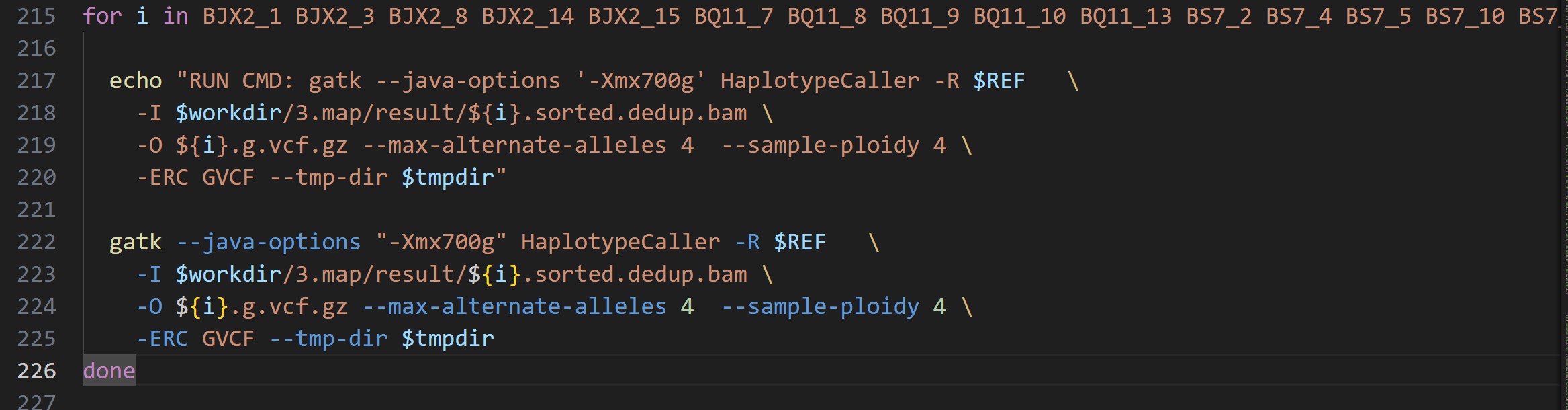
检查你的db导入是不是漏掉染色体了;
由于下游的分析软件很少有支持多倍体的vcf的,一般多倍体也当做2倍体分析;--sample-ploidy 2
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!