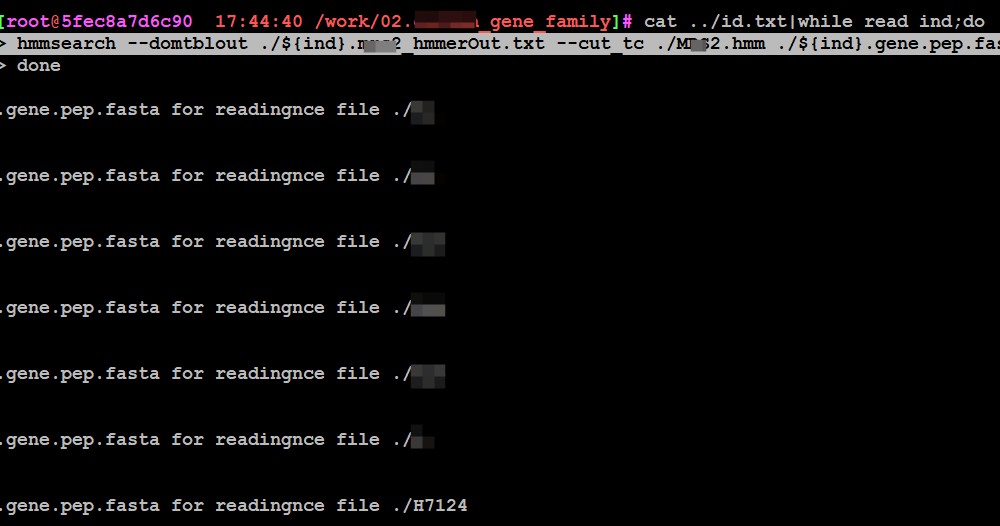
cat ../id.txt|while read ind;do
> hmmsearch --domtblout ./${ind}.2_hmmerOut.txt --cut_tc ./2.hmm ./${ind}.gene.pep.fasta
另外你在截图的时候图片尽量截全一点,报错信息全面一点方便分析问题
单独一个基因组执行的时候是没有问题的