你看下你的gvcf文件里面的染色体编号是不是4,还是chr4或者Chr4,是不一样的
重测序分析
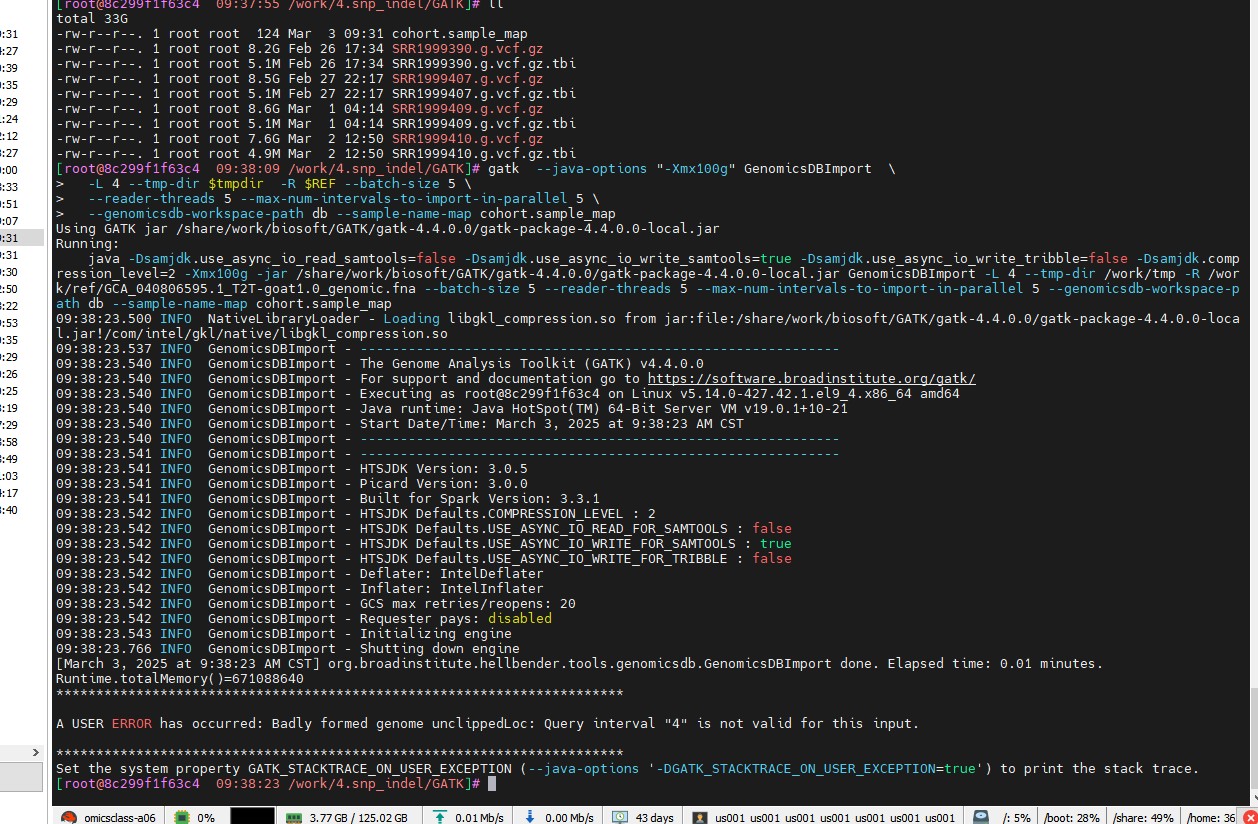
我进行重测序分析的时候,执行这个脚本的时候出现这个错误,命令为:
# ############合并GVCF方法2 大量样本 ########################## reseq:v2.0镜像支持,GATK 4.4.0.0
# gatk --java-options "-Xmx20g" GenomicsDBImport \
# -L 4 --tmp-dir $tmpdir -R $REF --batch-size 5 \
# --reader-threads 5 --max-num-intervals-to-import-in-parallel 5 \
# --genomicsdb-workspace-path db -V p1.g.vcf.gz -V p2.g.vcf.gz -V pool1.g.vcf.gz -V pool2.g.vcf.gz
#样本太多,可以输入列表
cd $workdir/4.snp_indel/GATK
#生成列表
ls *g.vcf.gz |while read a;do s=${a%%.g.vcf.gz} ;echo -e "$s\t$a";done >cohort.sample_map
#导入 db
gatk --java-options "-Xmx100g" GenomicsDBImport \
-L 4 --tmp-dir $tmpdir -R $REF --batch-size 5 \
--reader-threads 5 --max-num-intervals-to-import-in-parallel 5 \
--genomicsdb-workspace-path db --sample-name-map cohort.sample_map
#将gvcf转换成vcf
gatk --java-options "-Xmx20g" GenotypeGVCFs --tmp-dir $tmpdir -R $REF -O all.raw1.vcf.gz -V gendb://db
3 个回答
我比对后的gvcf文件染色体编号如下,能用这样的格式去制作chrlist 时候,命令显示不识别(A USER ERROR has occurred: Badly formed genome unclippedLoc: Query interval "chrlist" is not valid for this input.),这样的染色体编号是不是需要替换一下?
CP162209.1
CP162210.1
CP162211.1
CP162212.1
CP162213.1
CP162214.1
CP162215.1
CP162216.1
CP162217.1
CP162218.1
CP162219.1
CP162220.1
CP162221.1
CP162222.1
CP162223.1
CP162224.1
CP162225.1
CP162226.1
CP162227.1
CP162228.1
CP162229.1
CP162230.1
CP162231.1
CP162232.1
CP162233.1
CP162234.1
CP162235.1
CP162236.1
CP162237.1
CP162238.1
CP162239.1
擅长:重测序,遗传进化,转录组,GWAS
这个地方需要改一下,你的染色体编号有4吗? 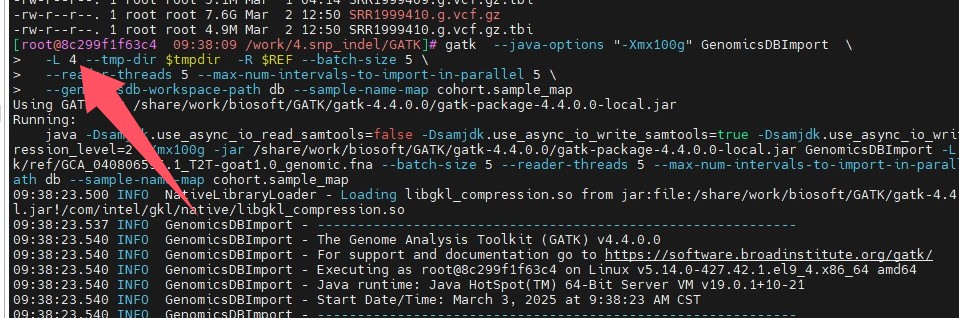
-L 也后面可以跟一个文件,里面一条染色体一行: chr.list
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
