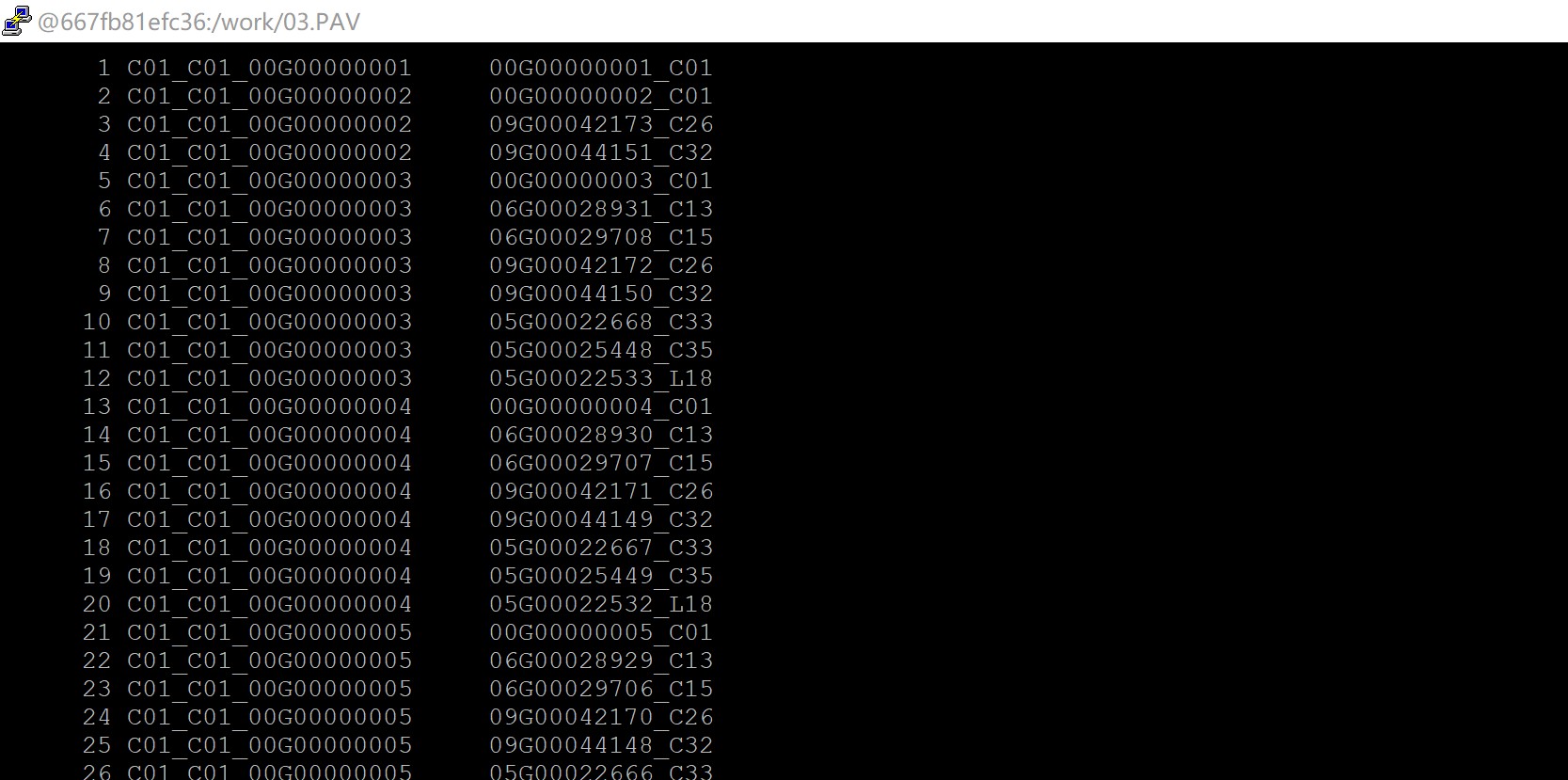
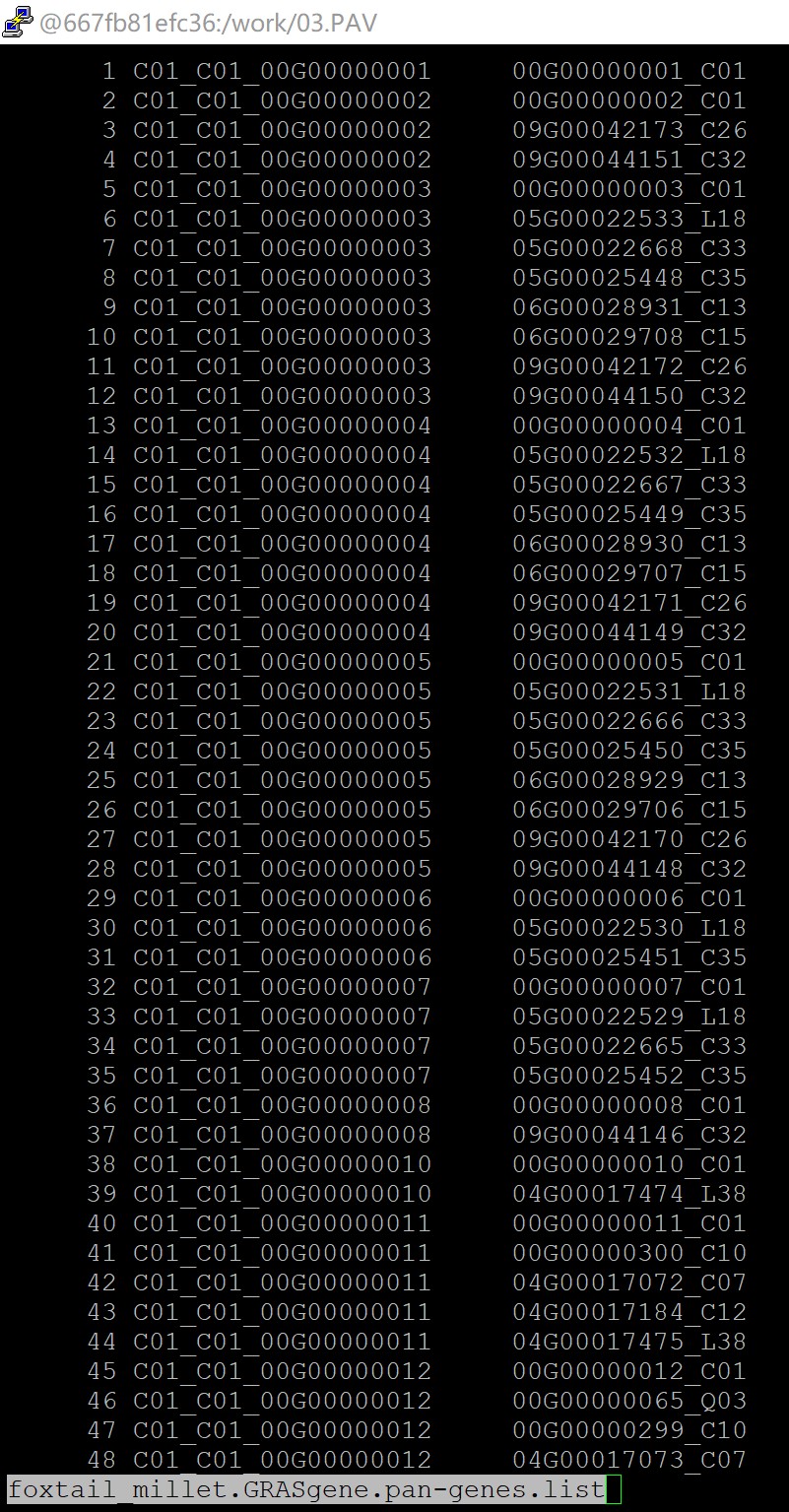
所以你在未做任何处理之前,你的gff文件里面基因ID的格式就是基因组名+基因id这样的是吗,我举个例子,比如说你的原始gff文件里面,你的基因编号就是C01_00G00000006对吗,要是你描述的这种情况,你需要把这条指令改成这样:awk 'FNR>1{print $1"\t"$3"_"$2"_"$3}' final.last.gene.pair.txt > yourfile.pan-genes.list
但是我们在做基因家族鉴定的时候,不是把基因组信息加在后面的吗,这是你自己的操作?我理解的如果你的原始基因id应该是这个C1_00G00000001,如果你按照我们的课程坐下来,你鉴定出来的基因id应该为:C1_00G00000001_C01,如果是你自己的行为你还是不要乱改脚本
还有你提到的这一点“之前是C01_C1_00G00000001,在做存在缺失时,我都把他们改了,我现在该怎么办啊”你不能随意的改动你原始的基因id,不然后面就会出现对不上的情况,你需要自己改回来,不然后面会出现很多类似的问题