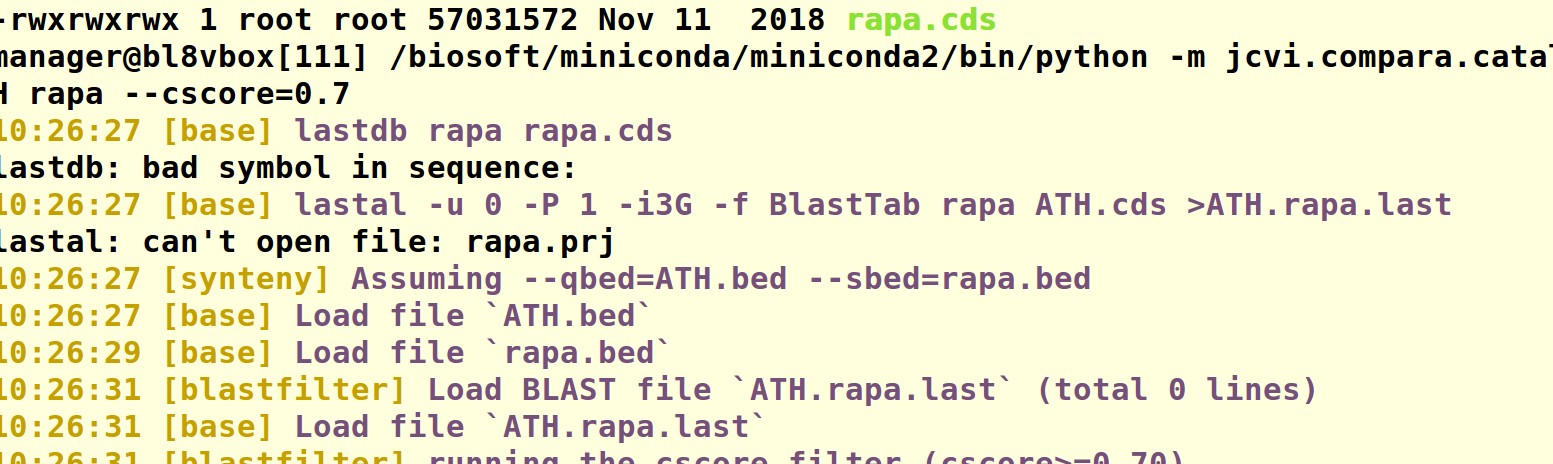
首先ll检查下你的当前路径下的文件,然后检查一下ATH.cds ATH.bed rapa.cds rapa.bed 这四个文件,或者截图我们帮你看一下。
5 老师,我是下载的最新版本的biolinux,但是在做不同物种之间的共线性分析比对时,最后出结果出图的时候出现这个了,请问怎么解决啊?
回答问题即可获得 10 经验值,回答被采纳后即可获得 10 金币。
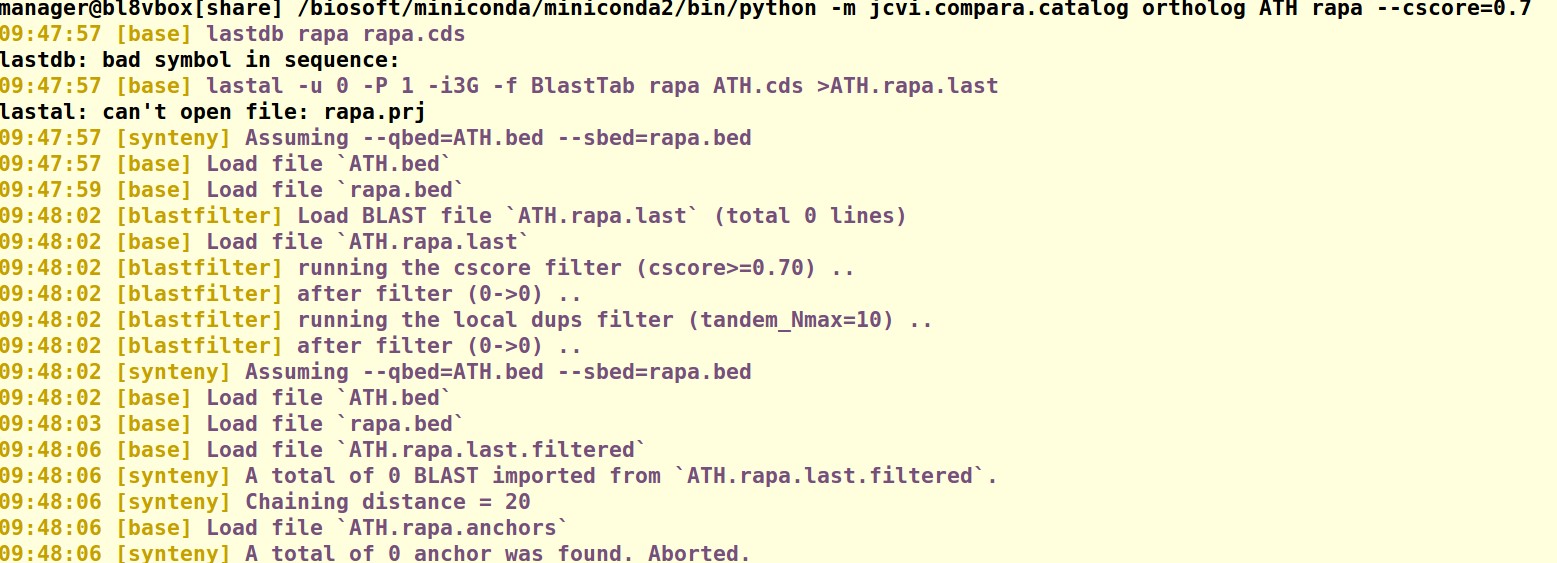
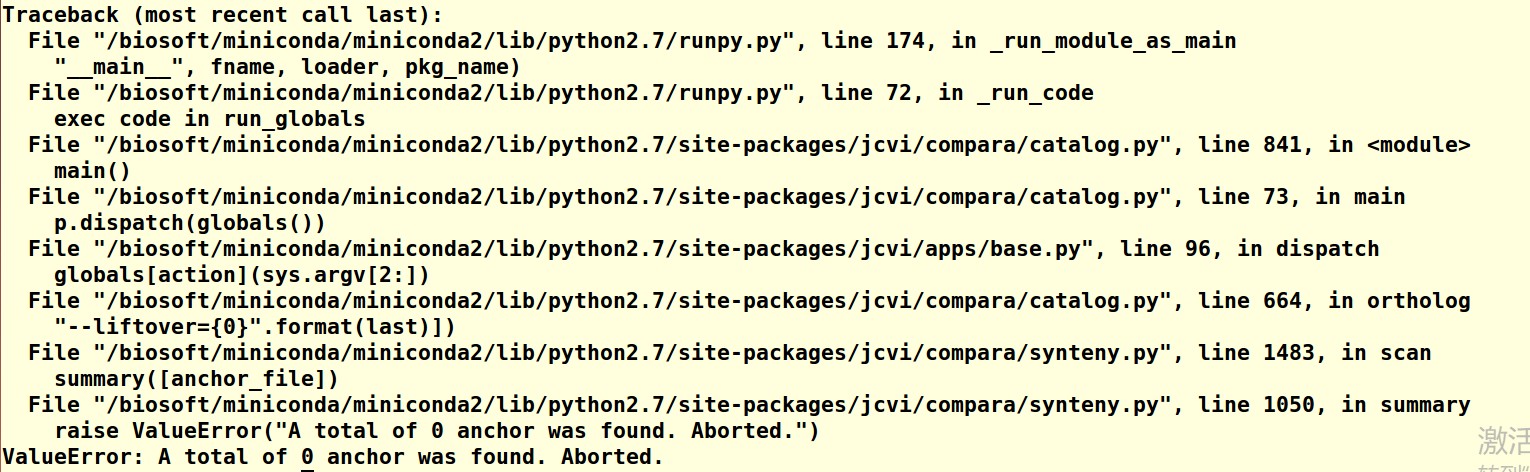 输入的命令为:/biosoft/miniconda/miniconda2/bin/python -m jcvi.compara.catalog ortholog ATH rapa --cscore=0.7。
输入的命令为:/biosoft/miniconda/miniconda2/bin/python -m jcvi.compara.catalog ortholog ATH rapa --cscore=0.7。
2 个回答
擅长:perl,基因家族,linux,chip-seq
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

老师,我都是用的原始的数据,在执行 /biosoft/miniconda/miniconda2/bin/python -m jcvi.compara.catalog ortholog ATH rapa --cscore=0.7 这条命令时,无法进行比对,没有生成 ATH.rapa.anchors 这个文件,导致后面的步骤无法进行;而且我是下载的最新版本的bioLINUX,麻烦老师帮忙看一下,谢谢老师。