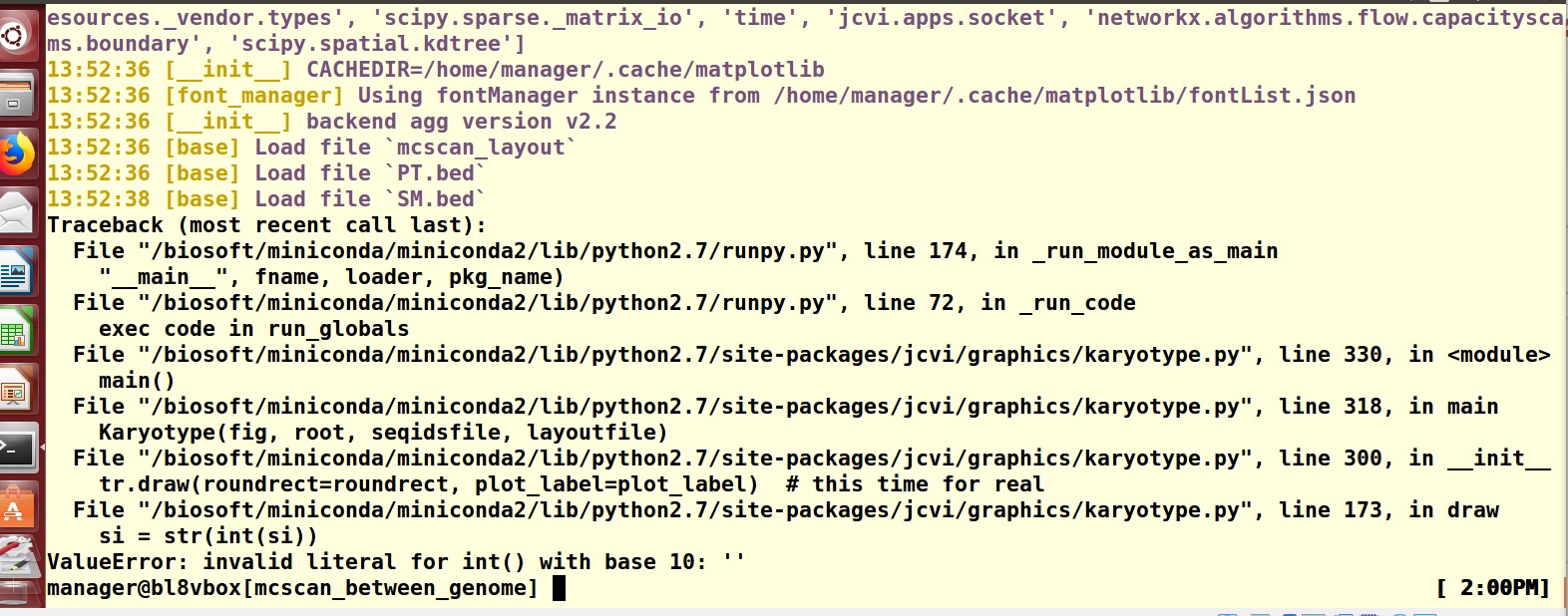
 这是有可能哪里出错了?
这是有可能哪里出错了? 以及SM.bed 都是对的,请问怎么解决?
以及SM.bed 都是对的,请问怎么解决?你再仔细检查一下你的输入文件是不是正确
不同物种之间的共线性分析
4 个回答

擅长:重测序,遗传进化,转录组,GWAS
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

谢谢,问题解决了
不同物种共线性分析时,出现以下报错信息怎么解决呢?no such file or directory /biosoft/miniconda/miniconda2/bin/python