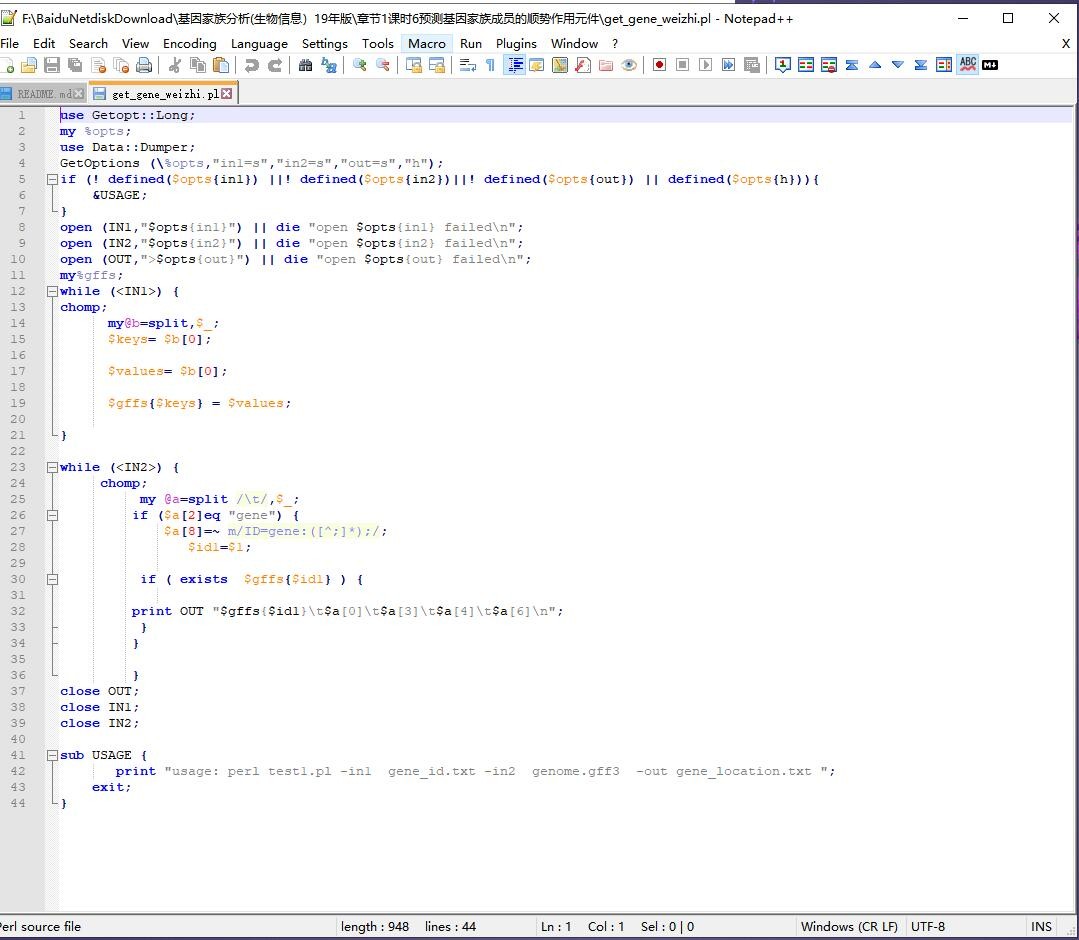
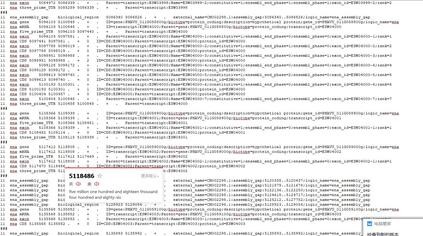
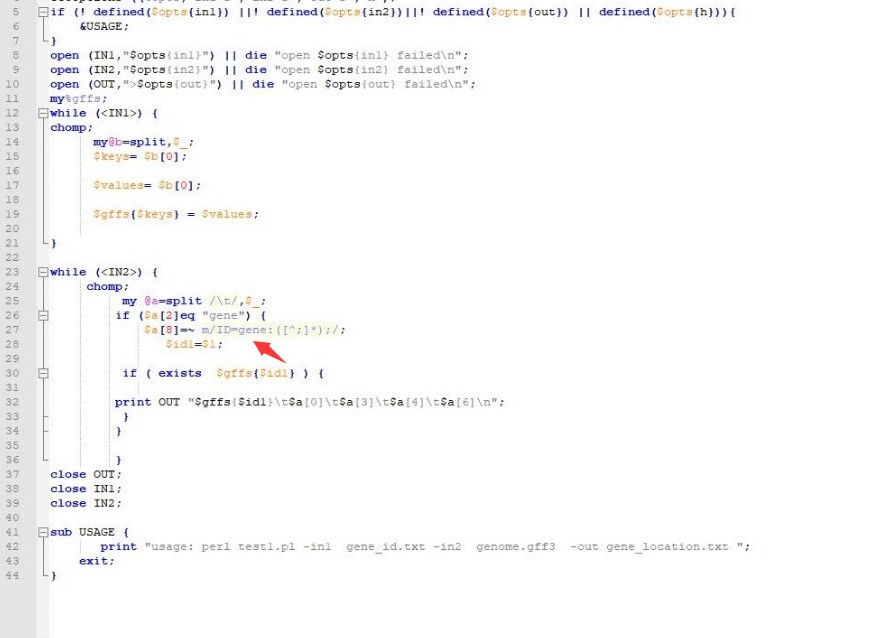
脚本认为ID=与;之间的为gene或mRNA的ID,建议你把GFF文件里面的gene: CDS: transcript: 删除; 然后再看看fasta里面的ID与GFF里面的ID是否一致。 这样保持所有地方的ID一致,就不会因为找不到对应的ID而出错。
如果上面按照我说的都改了,脚本这里27行就可以删除gene: 获取ID信息了:
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!