library(SummarizedExperiment)
library(dplyr)
library(DT)
library(WGCNA)
library(flashClust)
expr_file = "E:/MSC,ossu-9.1/1/WGCNA/data.txt"
trait_file = "E:/MSC,ossu-9.1/1/WGCNA/trait.txt"
workingDir = "E:/MSC,ossu-9.1/1/WGCNA"
setwd(workingDir)
#################################################### input data##########################
# The following setting is important, do not omit.
options(stringsAsFactors = FALSE)
#读取数据,删除第一列GeneSymbol
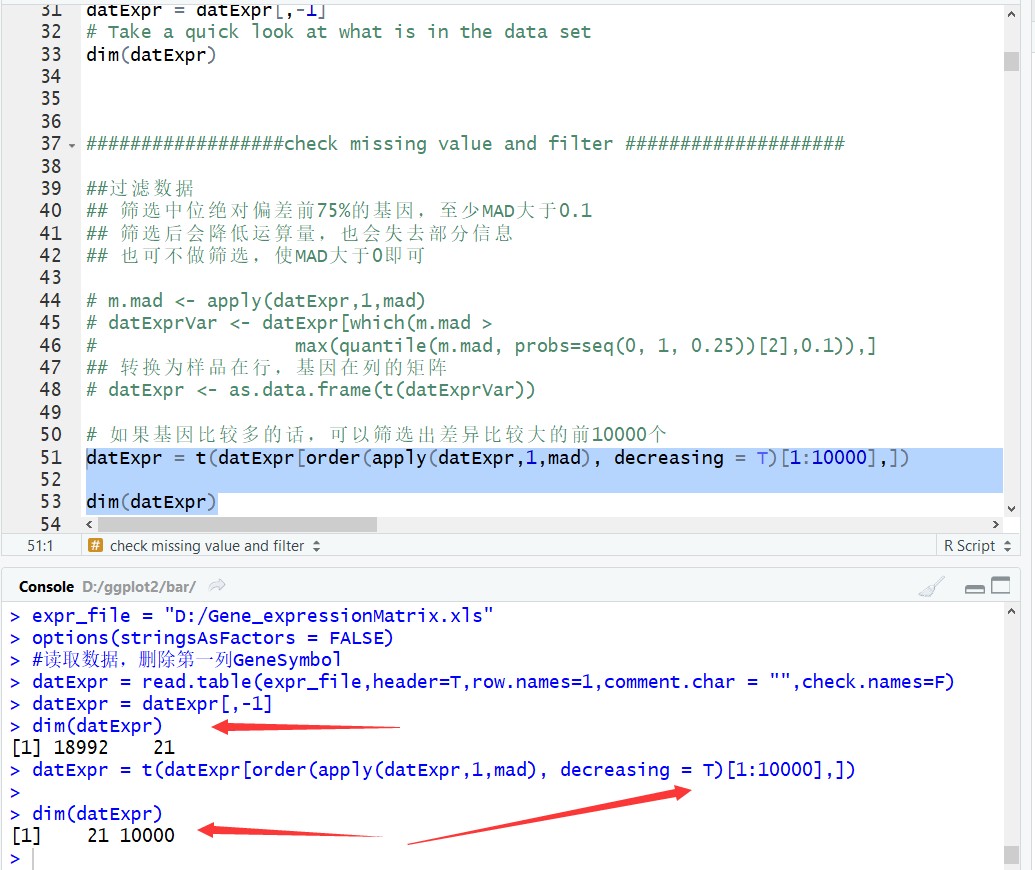
datExpr = read.table(expr_file,header=T,row.names=1,comment.char = "",check.names=F)
datExpr = datExpr[,-1]
# Take a quick look at what is in the data set
dim(datExpr)
##################check missing value and filter ###################
datExpr = t(datExpr[order(apply(datExpr,1,mad), decreasing = T)[1:5000],])
dim(datExpr)
根据上面的代码,我想把1.8w个基因缩减下,结果在进行MAD筛选前5k个基因的时候,出现,
> datExpr = t(datExpr[order(apply(datExpr,1,mad), decreasing = T)[1:5000],])
Error in x - center : non-numeric argument to binary operator
我查了下是什么“非数值参数为二进制运算符”,
不是太明白怎么回事。Genedata.xls
麻烦老师看看怎么回事
我后来发现,好像需要基因在行排列,那样可以不出现上面那个error,
fpkm = read.table("data.txt",header=T,comment.char = "",check.names=F)#########file name can be changed#####数据文件名,根据实际修改,如果工作路径不是实际数据路径,需要添加正确的数据路径
# Take a quick look at what is in the data set
dim(fpkm)
names(fpkm)
datExpr0 = as.data.frame(t(fpkm[,-1]))
names(datExpr0) = fpkm[,1];##########如果第一行不是ID命名,就写成fpkm[,1]
rownames(datExpr0) = names(fpkm[,-1])
##################check missing value and filter ####################
datExpr0
datExpr = t(datExpr0[order(apply(datExpr0,1,mad), decreasing = T)[1:5000],])
dim(datExpr)
不过后来我有碰到一个问题,形成一个大矩阵。。。。700+MB
然后,我用
datE <- as.data.fram(datExpr)
dim(datE)
[1] 18992 5000
我发现基因还是那么多个,和最开始的没有什么区别,唯一的区别只有GSM的序号变了位置。。没了啊。。不应该出来前5000吗
真是奇怪啊,这个代码。GEO那边演示也没有看到用什么格式的数据格式。。