有没有老师。。。
10 perl求助,基因ID的对应替换perl脚本怎么写
回答问题即可获得 10 经验值,回答被采纳后即可获得 15 金币。
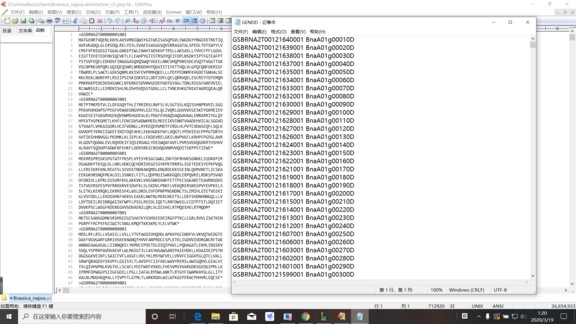 如图,想要将gene Alisa对应的Bna基因ID对应替换上去,perl脚本该怎么写。。
如图,想要将gene Alisa对应的Bna基因ID对应替换上去,perl脚本该怎么写。。
#!/usr/bin/perl ;
#usage:perl repalce.pl list.file fa.file out.file
open LIST,$ARGV[0];
open FAFILE,$ARGV[1];
open OUT,">$ARGV[2]";
undef $/;#
$in=<FAFILE>;#
study $in;
$/="\n";#
while(<LIST>){
chomp;
my($new,$old)=split;
$in=~s/$old/$new/;
}
这个是网上的perl,out文件是空白的。。求老师看一下
3 个回答
擅长:重测序,遗传进化,转录组,GWAS
这个问题可以写个perl脚本或者python脚本完成,分别用bioperl或者biopython包处理fasta文件会很方便,有相应课程;
不用bioperl 给你个写脚本思路:首先把ID的对应关系读到一个hash中,然后判断只接处理ID行,也就是带有>的行,其他行输出就行;
生物信息入门到精通必修基础课:、perl语言高级、python语言入门到精通
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

#!/usr/bin/perl
# Filename: repalce.pl
# Description:replace the file ID.
# Author:xueqc_
# Vision: 1.12
# Date: 2020.03.21
# Usage:perl repalce.pl list.file fa.file out.file
use warnings;
use strict;
my @Gene_Alisa_ID;
my $i="";
open LIST,"<$ARGV[0]" or die("Can not open the file!$!");
while(<LIST>){
my $line=$_;
if($line=~/^\s*\n/){next;}
$line=~s/\r//;
$i.=$line;
}close LIST;
$i=~s/\s+/\t/;
@Gene_Alisa_ID=split(/\s/,$i);
open FA,"<$ARGV[1]";
open OUT,">$ARGV[2]";
while(<FA>){
my $line=$_;
$line=~s/\r?\n//;
if($line=~s/^>(.+)\s*/$+/){
$i=0;
while($i<@Gene_Alisa_ID){
if($line eq $Gene_Alisa_ID[$i]){
$line=~s/$Gene_Alisa_ID[$i]/$Gene_Alisa_ID[$i+1]/;
$Gene_Alisa_ID[@Gene_Alisa_ID]=$Gene_Alisa_ID[$i];
$Gene_Alisa_ID[@Gene_Alisa_ID]=$Gene_Alisa_ID[$i+1];
splice(@Gene_Alisa_ID,$i,2);#将匹配了的LIST_ID对排到数组的最后
last;
}elsif((length($line)>length($Gene_Alisa_ID[$i])) and $line=~s/$Gene_Alisa_ID[$i]\s.*/$Gene_Alisa_ID[$i+1]/){#如果fa文件ID号后有注释的话,将被删掉
$Gene_Alisa_ID[@Gene_Alisa_ID]=$Gene_Alisa_ID[$i];
$Gene_Alisa_ID[@Gene_Alisa_ID]=$Gene_Alisa_ID[$i+1];
splice(@Gene_Alisa_ID,$i,2);#将匹配了的LIST_ID对排到数组的最后
last;
}else{
$i+=2;
}
}
print OUT ">$line\n";
if($i == @Gene_Alisa_ID){
$line=~s/\s.+//;
print "The gene alisa list is missing ID: $line\n";
}
}else{print OUT $line."\n";}
}
close FA;
close OUT;