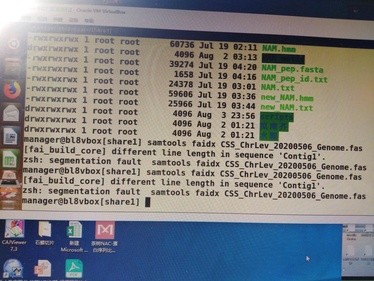
你的文件有问题你再处理一下你的文件格式看看是不是出错了;
序列行每一行的长度应该一致
你可以用get_fa_by_id.pl 脚本重新提取一下基因组里面的所有序列文件;https://www.omicsclass.com/article/179
处理数据还是要学习编程语言的:
你的文件有问题你再处理一下你的文件格式看看是不是出错了;
序列行每一行的长度应该一致
你可以用get_fa_by_id.pl 脚本重新提取一下基因组里面的所有序列文件;https://www.omicsclass.com/article/179
处理数据还是要学习编程语言的:
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
