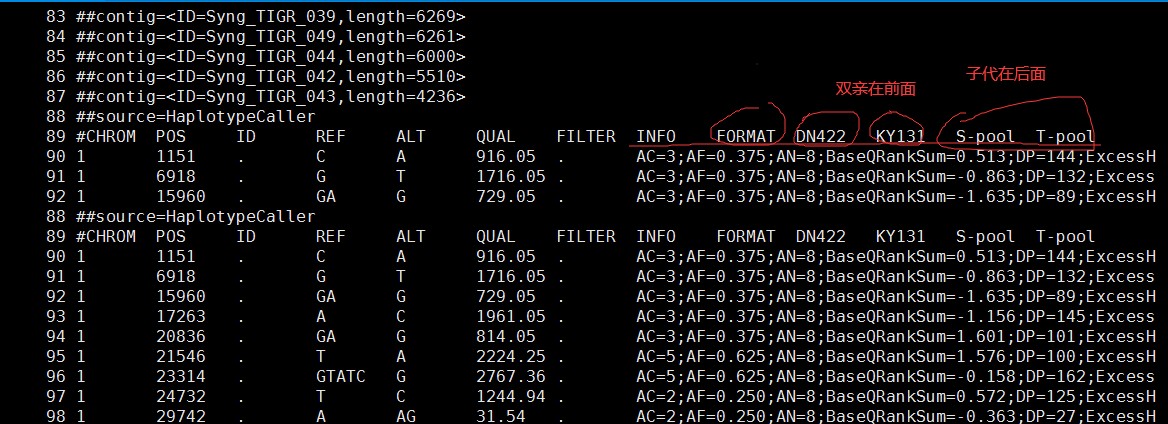
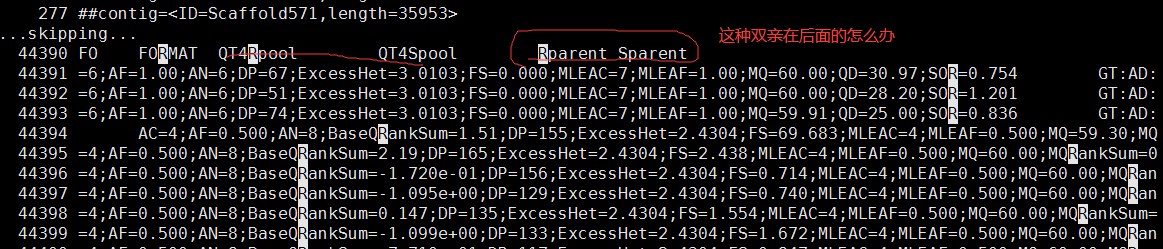
老师在课程中使用的示例文件中vcf文件中四个样本的顺序是双亲,然后子代。但是我发现我的vcf文件的顺序是双亲在后面,两个子代池在前面,这种怎么调整顺序呢,或者会不会影响qtlseq的运行
如果用的是组学大讲堂提供的docker镜像就不需要更改,只是命令行参数样本名字写对就可以,因为我们对qtlseq这个python脚本做过修改;
但是,你自己安装的软件就按照软件说明使用。
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!