
 图三
图三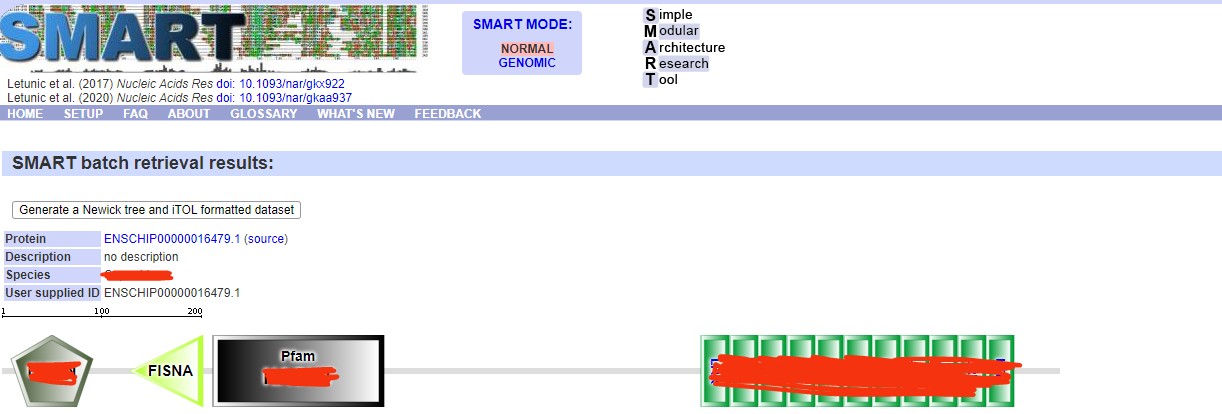
人类的基因 基因家族基因都是明确的你直接查询就行了;不用鉴定
BLAST 比对建库
2 个回答

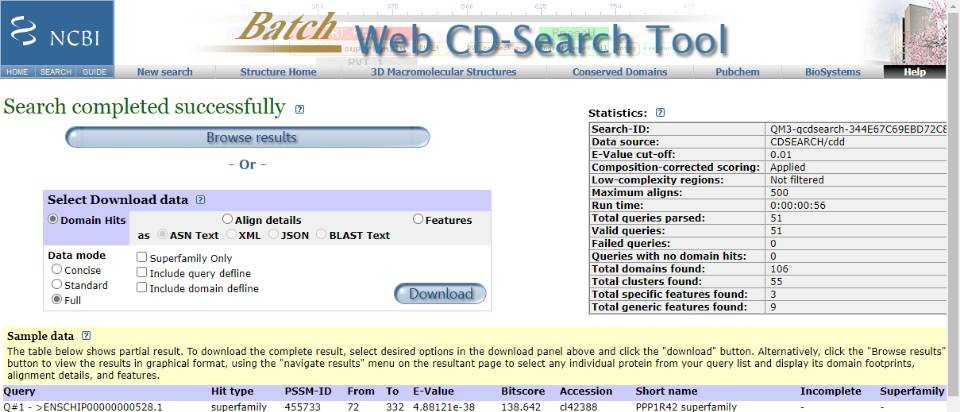
老师,您误会了。因为我的目标物种没有pfam号,所以我用已知人类的蛋白序列建立种子序列,分析结果如上所示。通过前期的文献积累,比对结果符合该基因家族的特征。
目前的主要困惑是:(1)在CD-search 里面,Incomplete出现N/C/NC端不完整的片段,是直接从XXXX.fa里面删除整条ID对应的序列还是保留分析?(2)在完成三大数据库的比对后,在hmmsearch第一次搜素结构域时,我如何利用比对结果建立物种特异hmm模型?