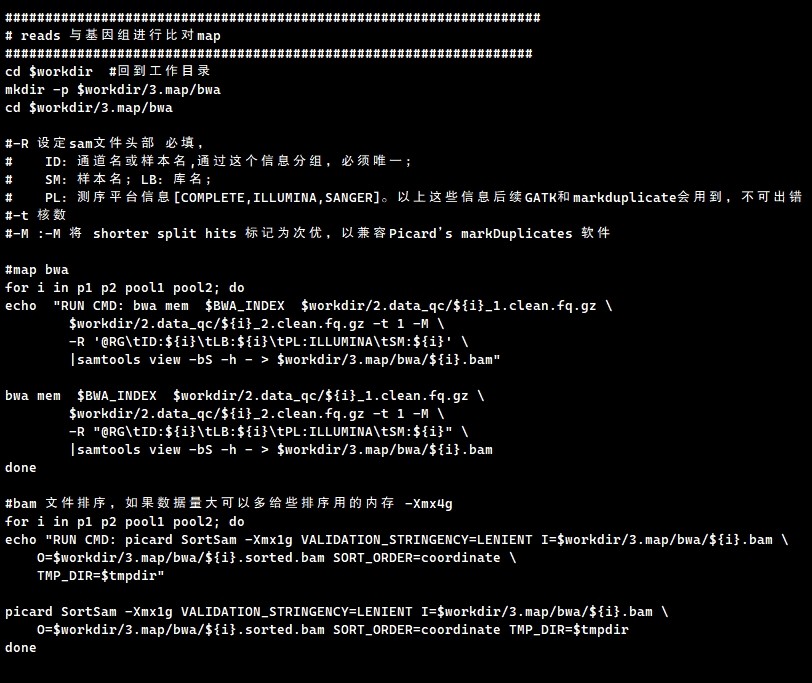
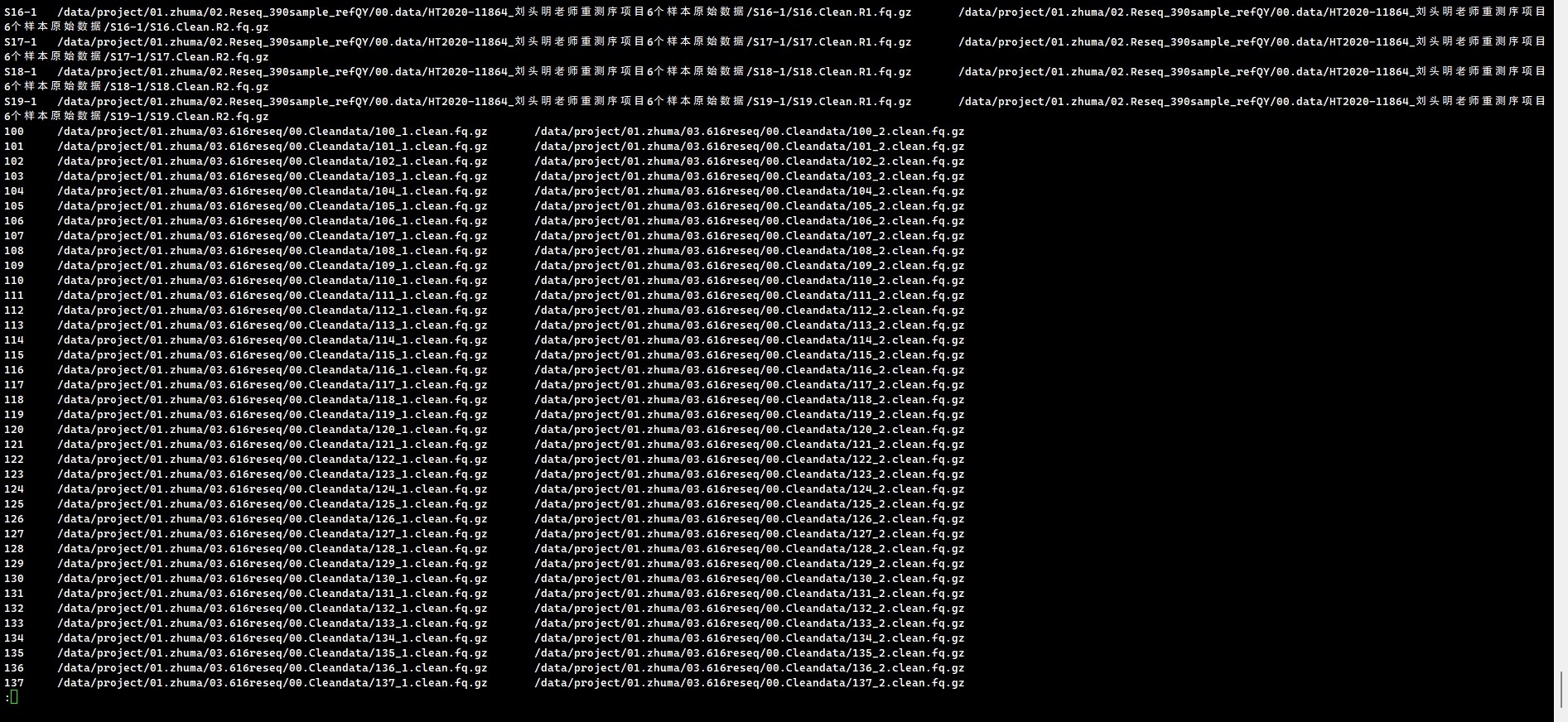
可以整理一个样本数据文件列表,第一列为样本ID,第二列为fq1的文件绝对路径,第三列为fq2的文件绝对路径;把for循环替换成while 循环;
例如:
cat data-path-list.txt | while read i fq1 fq2;do
bwa mem $BWA_INDEX $fq1 $fq2 -t 1 -M \
-R "@RG\tID:${i}\tLB:${i}\tPL:ILLUMINA\tSM:${i}" \
|samtools view -bS -h - > $workdir/3.map/bwa/${i}.bam
done
