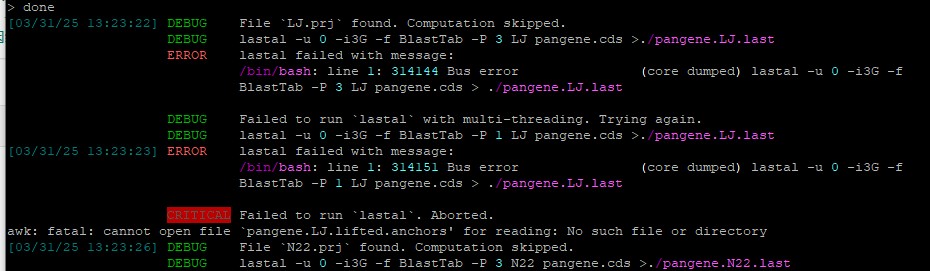
last报错应该是因为内存不足:https://www.omicsclass.com/article/1413
泛基因集构建中JCVI做共线性分析构建完一半数据报错 core dumped
for i in `cat stepmap_ID.txt`;do
# JCVI做共线性分析,--cpus 5 限制计算资源使用,如果不加默认--cpus 56
python3 -m jcvi.compara.catalog ortholog \
pangene ${i} --cpus 5 --no_strip_names --cscore 0.8;
# 以提升后的anchors作为基因对的结果,把比对上的基因排序去重输出到mapped.txt文件
anchor=pangene.${i}.lifted.anchors;
awk '{ print $2}' $anchor | sort -u > ${i}.mapped.txt;
# 未能比对上的基因合并到模板里,组成新的模板,再和下一个基因组比对
awk '{if(ARGIND==1) {val[$0]}else{if(!($4 in val)) print $0}}' ${i}.mapped.txt ${i}.bed >> pangene.bed ;
awk '{if(ARGIND==1) {val[$0]}else{if(!($4 in val)) print $4}}' ${i}.mapped.txt ${i}.bed |seqkit grep -f - ${i}.cds >> pangene.cds;
done