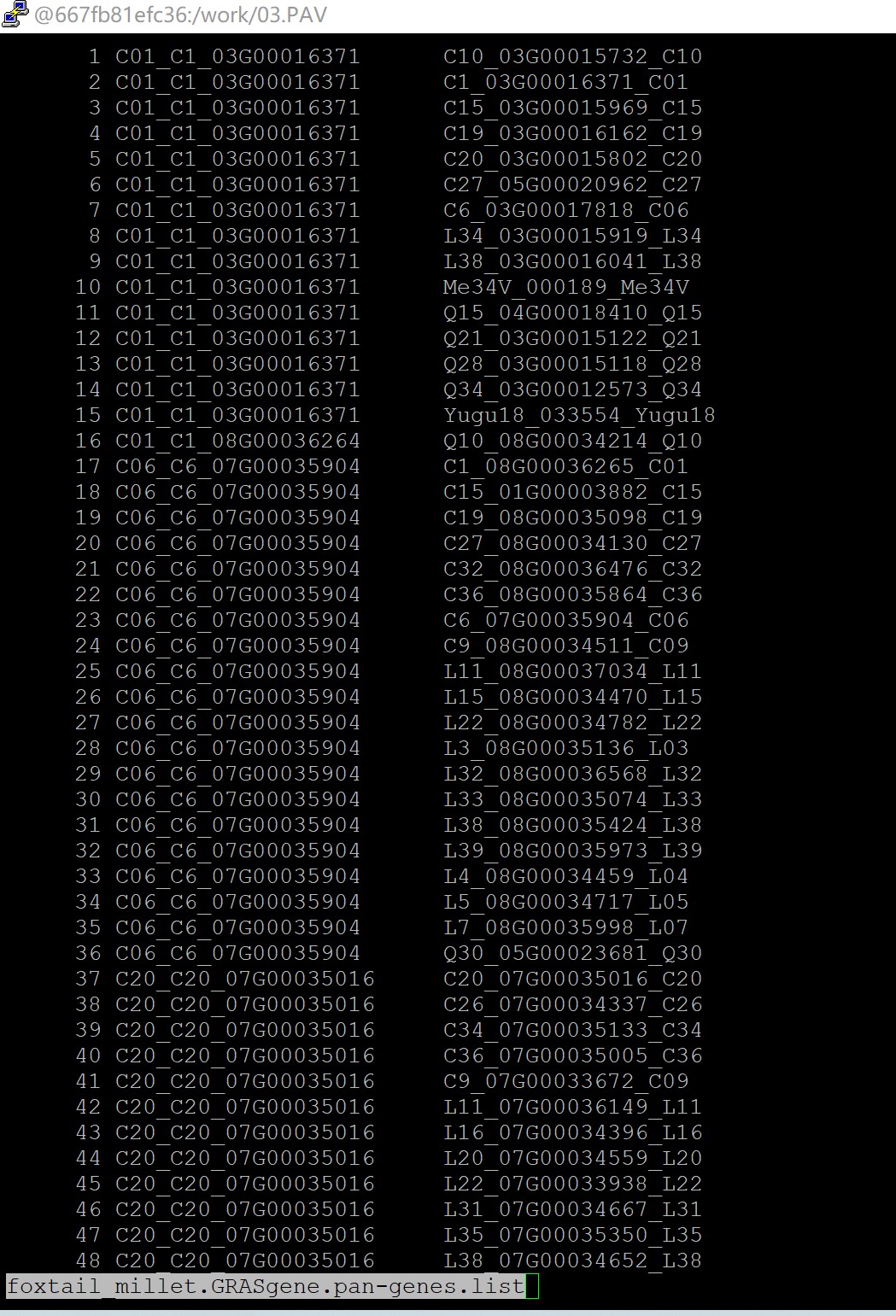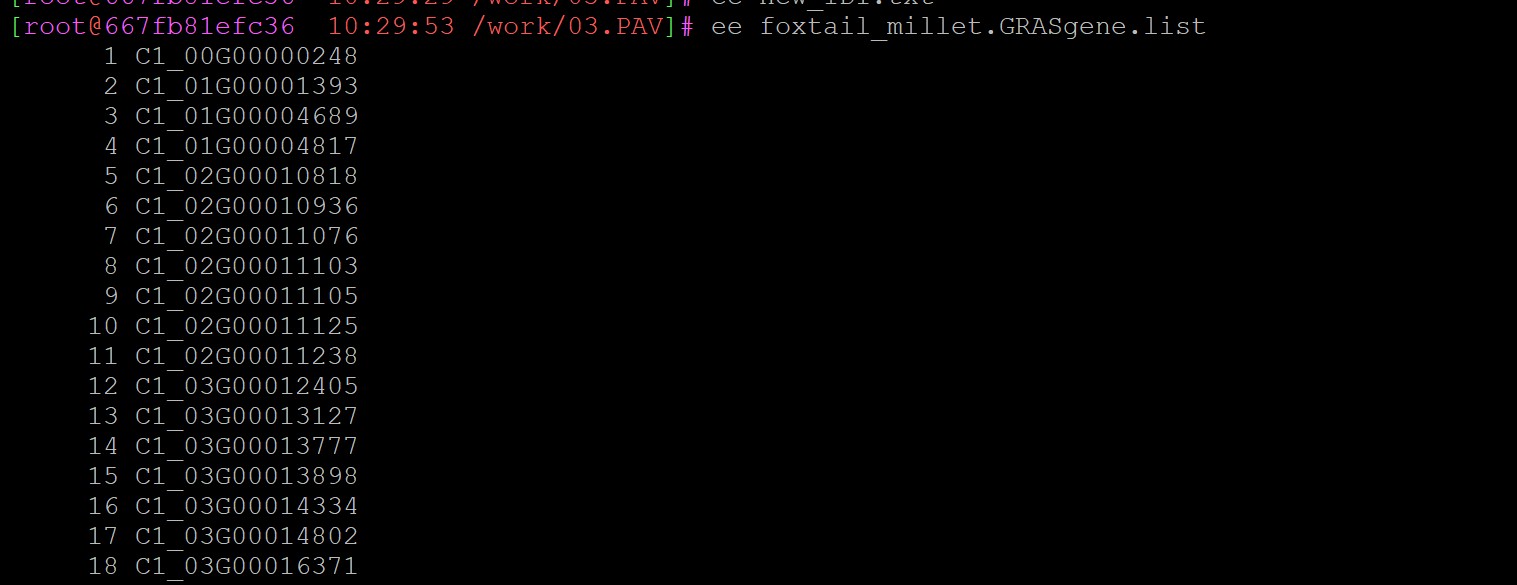
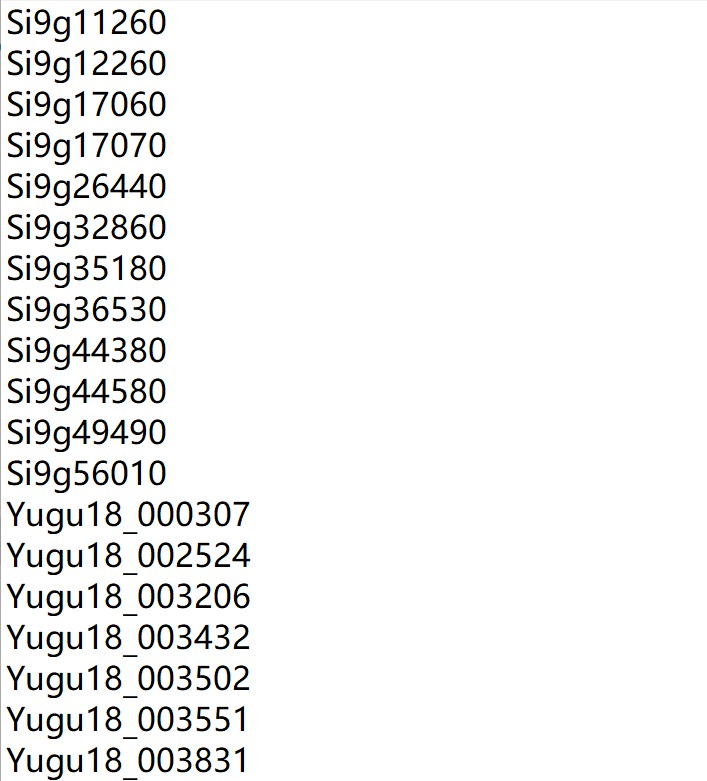

 C01对应的原始GFF文件里基因名就是C1_00G000006,xiaomi对应的基因名是Si9g11260,Yugu1_T2T.genome对应的基因名是Seita.9G123000_Yugu1_T2T.genome。 (我要修改ID.txt文件吗?)
C01对应的原始GFF文件里基因名就是C1_00G000006,xiaomi对应的基因名是Si9g11260,Yugu1_T2T.genome对应的基因名是Seita.9G123000_Yugu1_T2T.genome。 (我要修改ID.txt文件吗?)同学,其实前面已经给你说过解决办法了,最好的办法就是你重新跑流程,不要乱改你的基因id,你改了之后我们也很难改流程去对你的数据进行调试,你可以把构建泛基因列表的代码这里改一下,然后回到你修改id之前重新跑一下,总之不要改动你原始的基因id,不然后面对应起来就是很麻烦,你按照流程跑:
# 把第二列的前缀去掉
awk -F'\t' 'BEGIN{OFS="\t"} { if (match($2, /_/)) {$2 = substr($2, RSTART+1)};print}' last.gene.pair.txt|\
sed "s/ /\t/g" | sed "1i\PangeneID\\tgeneID\\tSpieces\\tChrom\\tStart\\tEnd" > final.last.gene.pair.txt