15 老师;我这儿GFF3文件格式和教学视频不一样;蛋白序列上显示gene_id有三个;我不知道如何选取哪一个进行接下来的GFF3基因位置分析;GFF3文件上没有染色体位置信息,和教学视频的格式不同-gene出现在第3列;gene_id出现在第9列我该如何选取有效的ID呢?如何修改Perl脚本?如何能成功提取基因位置信息呢?望老师指点!谢谢呢
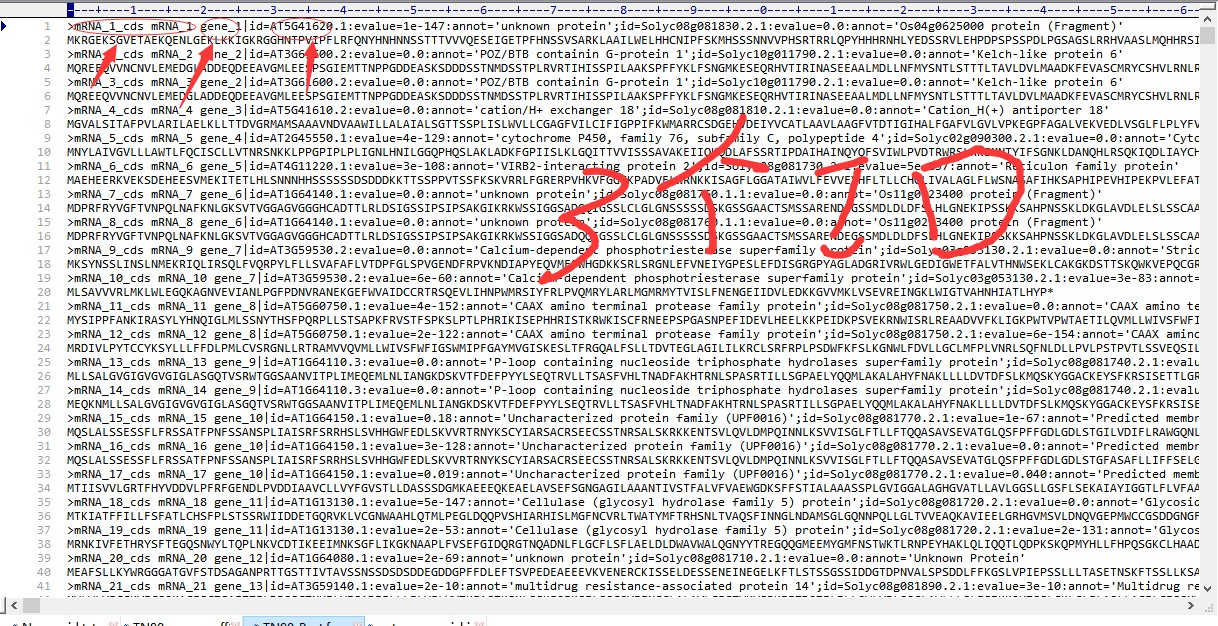



- 老师;我这儿GFF3文件格式和教学视频不一样;蛋白序列上显示gene_id有三个;我不知道如何选取哪一个进行接下来的GFF3基因位置分析;GFF3文件上没有染色体位置信息,和教学视频的格式不同-gene出现在第3列;gene_id出现在第9列我该如何选取有效的ID呢?如何修改Perl脚本?如何能成功提取基因位置信息呢?望老师指点!谢谢呢
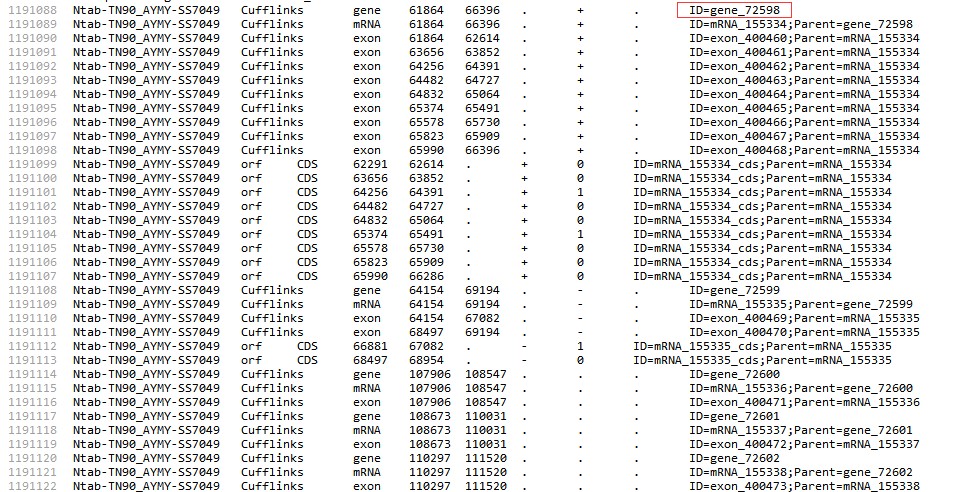
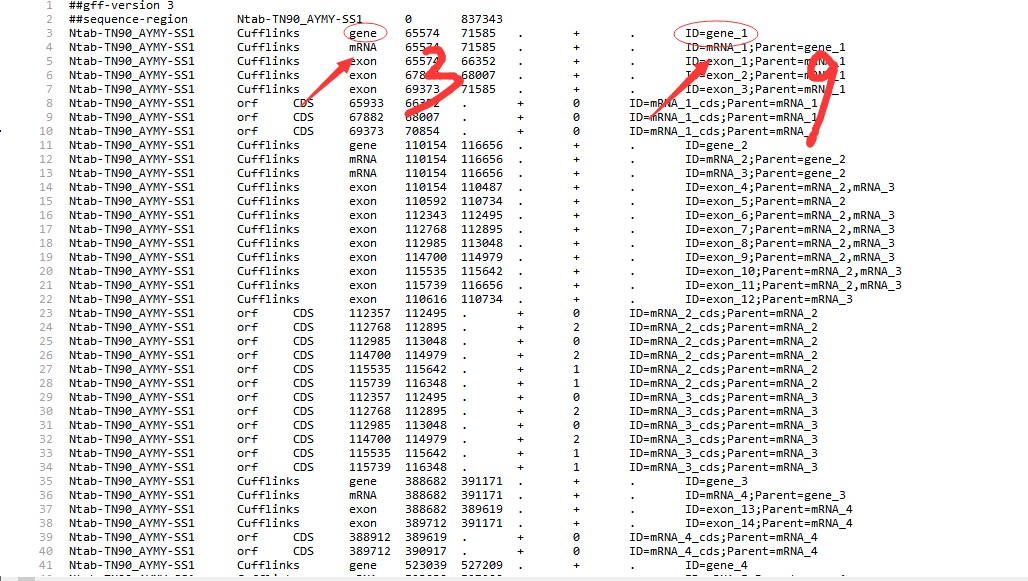
 老师这个GFF3文件的Id和之前用Perl提取出来的ID对不上呢?
老师这个GFF3文件的Id和之前用Perl提取出来的ID对不上呢?其它 2 个回答

1.fasta文件,第一个空格前面才是序列ID,后面的都是序列的注释信息,我们的脚本会忽略注释信息,只读ID
2.GFF文件,gff文件,我们的脚本只会读ID= 后面的ID信息;其他的信息会忽略,你贴的脚本很多,不知道你的问题是哪一个?
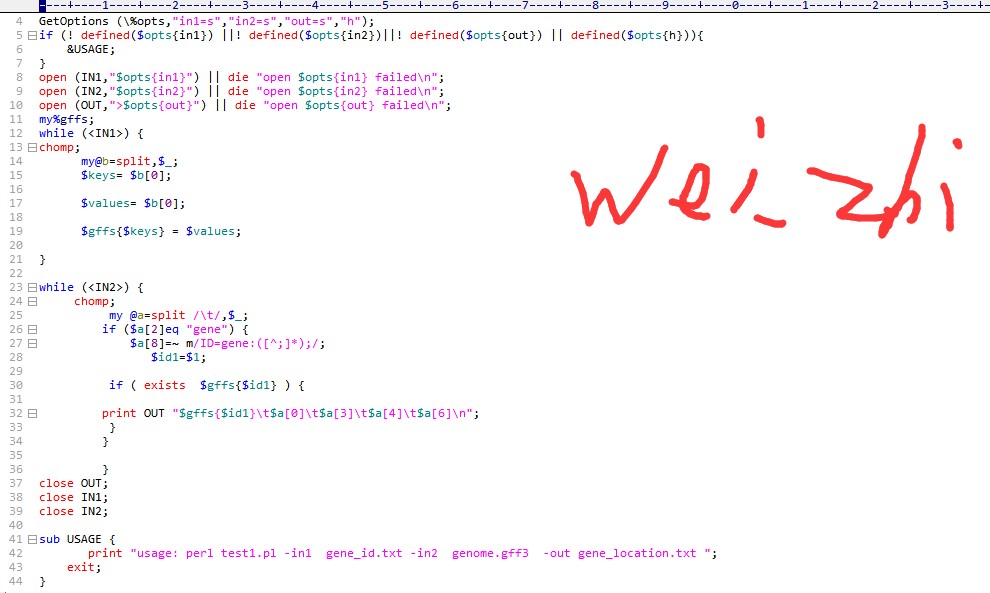
3.运行位置脚本的时候,ID要对应才能得到正确的结果,所以要观察自己的文件是否ID匹配;看你的这个脚本里面添加了gene:,你需要修改一下这里,视频里面应该也提示过:
建议学习一下perl语言,perl入门到精通、perl语言高级
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
